
Abstract
While mostly diagnosed in the pediatric population, neurological complications of pandemic influenza A infection may affect young and previously healthy adults, and may follow a fulminant, severe, and occasionally fatal course. We reviewed severe neurological complications secondary to influenza infection reported in the literature, in attempt to outline recurrent syndromes that may assist the clinician in making a timely diagnosis. Vigilance and awareness of these clinical entities are key in the neurologist and intensivist’s role in surveillance and early recognition of pandemic influenza, and in ensuring improved survival for affected patients.
Similar content being viewed by others

Introduction
Influenza-associated neurological complications encompass a wide range of clinico-pathological entities. Many have been reported and are well recognized. Clinical manifestations may affect any level of the neuraxis, and present with a large spectrum of severity, ranging from a mild and progressive course with full symptom resolution to an aggressive disease rapidly culminating in fatality [6, 13, 39, 44, 56, 93]. While most cases reported pertain to the pediatric population, a surge in the incidence of severe diseases in young and otherwise healthy adults is noted during pandemics. Emergency preparedness is necessary as the medical community remains on the lookout for a novel influenza pandemic. The neurologist and neurological intensivist have a role in epidemic surveillance and, therefore, should be aware of influenza-associated neurological complications to correctly and timely substantiate the diagnosis.
Review of available data in the medical literature on pandemics of different local and international impact from the last century shows a recurrent pattern of clinical presentations. We aim to review these clinical entities and current theories on their pathophysiology, and to outline a diagnostic and therapeutic approach to suspected cases of influenza-associated neurological complications.
Methods
We have conducted a Medline search of the scientific literature using the MeSH terms “influenza”, “influenza vaccine” or subtypes from recent pandemics (“H1N1”, “H5N1”, “H3N2”, “H2N2”). These results were matched with different MeSH terms, such as “encephalopathy”, “meningitis”, “myelitis”, “Guillain-Barré syndrome”, “myositis”, “neuritis”, or “encephalomyelitis.” Limits were placed to identify articles with abstract or full-text available in English or French. We reviewed all case reports, laboratory studies and past reviews pertaining to complications associated with influenza A infection and vaccination.
Clinical Presentations Associated with Influenza Infections
Over the past century, several subtypes of influenza A have caused pandemics with different impacts worldwide. The “Spanish flu” of 1918–1919 has been the single most fatal pandemic, caused by an H1N1 subtype of influenza A. An important epidemiological feature is the relative absence of influenza-associated neurological complications in the adult population outside of pandemics [51]. Sporadic cases have been reported from seasonal influenza, predominantly affecting the pediatric population [4, 44].
The Early 1900s
Historically, potential severity of a worldwide influenza pandemic surfaced following two closely occurring epidemics circa 1900. In particular, the H1N1 pandemic of 1918–19 has received much attention due to its geographical spread and high mortality. Two fatal clinical and pathological entities have been associated with the epidemics of the early 1900s: Strümpell-Leichtenstern’s acute hemorrhagic influenza encephalitis (SLE), and von Economo’s encephalitis lethargica (EL) [21, 22, 55, 76].
Von Strümpell and Leichtenstern simultaneously defined SLE in detail at the end of the 19th century [22]. Clinically, it mostly affected children, and presented as somnolence, stupor or coma, with possible seizures and paralysis, over a course of days to weeks. Outcomes ranged from complete recovery to varying degrees of psychiatric or motor sequelae [21]. Pathologically, it was described as a cortical and basal ganglia hemorrhagic encephalitis, with pial infiltration and possible venous sinus thrombosis. Brainstem and cerebellum were occasionally affected [21, 22]. However, it was uncommon in the German epidemic of the 1890s, when it was first reported, and was reported at an even lower incidence in the Spanish flu of 1918–19 [21, 76]. Nonetheless, it bears similarities with influenza-associated encephalopathy syndromes subsequently reported over the 20th century. [21].
On the other front, much debate surrounded von Economo’s EL since the end of the 1918–19 pandemic [55, 59, 76]. While thousands of deaths have been ascribed to EL, its causal relationship to influenza infection has never been proven. EL was diagnosed over a wide timeframe: the first cases over 2 years prior to the influenza pandemic, and the last, up to 10 years after [21, 76]. It includes three different clinical subtypes affecting mainly young and previously healthy adults, with disturbance of sensorium and sleep patterns, and with or without parkinsonian features [21, 76]. Well-described pathological features showed significantly less inflammation when contrasted with SLE. Initially believed to be secondary to a highly neurotropic virus, its causal agent has failed to be identified, even after re-examination of exhumed patient tissues [22, 55, 76]. The difficulty in establishing an etiology, its fatality and its disappearance since the 1920s have cast into doubt its definition and existence over the past decades [22, 59, 76].
Influenza-Associated Complications: Data From Seasonal Influenza and Recent Epidemics
Since the pandemic of 1918–19, several other epidemics with smaller repercussion have surged. Virulent strains isolated include the H1N1, H3N2, and the H5N1 subtypes [13, 14, 24, 28, 33, 81]. Meanwhile, several clinical syndromes surfaced as recognizable central nervous system complications of the infection. Most reported cases originate from Japan and Taiwan, where the annual incidence of influenza-associated neurological complications are disproportionately elevated compared to other areas of the world. Much of the experience is drawn from the pediatric population, up to 5 years of age [33, 46, 67]. While preponderance for developing these complications are not exclusive to either East Asians, children, and the elderly, individuals with pre-existing neurological or neuromuscular diseases are also at increased risk [4, 28, 39, 63].
Most commonly encountered clinical syndromes can be classified into five subtypes of influenza-associated encephalopathy [57, 83]. Other presentations have been described, such as influenza-associated myopathy, abnormal movements suspicious for extrapyramidal involvement, transverse myelitis, Guillain-Barré syndrome, and post-infectious acute disseminated encephalomyelitis [52, 77, 85]. Of interest, as pointed out by Drago et al [16]., viral meningoencephalitis has only infrequently proven causal association with influenza infection.
Of the above-stated clinical entities, influenza-associated encephalopathy (IAE) has been most frequently reported to lead to fatal outcome. IAE is clinically defined as a group of syndromes with various degrees of cerebral dysfunction closely following a documented influenza infection, ranging between 1 and 14 days after the first systemic symptoms, with a median of 3 days [4]. Pathologically these syndromes span a large spectrum of severity but commonly show no signs of inflammation on cerebrospinal fluid or tissue examination [28]. Several subtypes of IAE have been described through case reports and various reviews in attempt to devise a formal classification of their clinical and laboratory profiles. In 2007, Mizuguchi et al [57] reviewed acute encephalopathy syndromes associated with several viruses, including influenza, and proposed a classification system by suspected pathophysiology. In 2009, Takanashi recognized two benign forms of IAE [86]. In 2010, Akins et al [3]. further described subtypes of acute encephalopathy with malignant cerebral edema. No consensus currently incorporates all reported influenza-associated encephalopathy syndromes described in the literature. Meanwhile, it is important to recognize that, although these clinical syndromes are most commonly associated with influenza infection, they are not exclusively the result of influenza disease: systemic responses to other viral or bacterial infections may lead to similar clinical presentations [4].
Major syndromes and classifications described are listed in Table 1. In the present review, we will revisit major IAE syndromes described in order of severity of illness. Mild encephalopathy with reversible splenial lesion (MERS) is a variant of acute encephalopathy with favorable outcomes. Encephalopathy with malignant brain edema (EMBE), hemorrhagic shock and encephalopathy syndrome (HSES) and acute necrotizing encephalopathy (ANE) are often rapidly progressive variants with diffuse cerebral dysfunction and poorer outcomes. Acute encephalopathy with seizure and late restricted diffusion (AESD) encompasses two variants of IAE with focal cortical edema often, yet again not exclusively, associated with influenza infection: acute infantile encephalopathy predominantly affecting the frontal lobes (AIEF) and hemiconvulsion-hemiplegia syndrome (HHS).
Mild Encephalopathy with Reversible Splenial Lesion
MERS has been described in numerous case reports and series [3, 20, 25, 87]. It is clinically characterized by a systemic prodrome of influenza infection with fever, vomiting, diarrhea, and cough, followed by the onset of delirium, decreased level of consciousness or seizures within one to 3 days. Investigations may reveal mild CSF pleocytosis, diffuse slowing on electroencephalogram (EEG) and a midline diffusion-restricting lesion in the splenium of the corpus callosum in an otherwise normal MRI [20, 88]. The splenial lesion is speculated to be reflective of intramyelinic edema with fiber layer separation [25]. Within a month, the patient fully returns to baseline clinical status with complete resolution of EEG and MRI findings, independently of treatment attempts. No therapies have been shown to alter the natural history of the disease or its symptomatology [20].
In 41 % of reported cases of MERS, no infectious agent is identified. Among the remaining cases, most commonly reported pathogens are influenza (A or B strains, 19 %), followed by mumps, adenovirus, rotavirus, varicella zoster virus, streptococcal, Staphylococcus aureus, Escherichia coli O-157, and Legionella pneumophila infections [25, 90]. When compared to other pathogens, influenza-associated MERS has a higher incidence of delirium; other manifestations of altered mental status reported include frontal lobe dysfunction, mild disturbance in level of consciousness (35 %) and seizures (33 %) [87].
Encephalopathy with Malignant Brain Edema
EMBE is also referred to as “acute brain swelling” by several authors, and Akins et al. have proposed the more clinically discriminate term EMBE in 2010. Fewer cases have been reported in the literature when compared to the other IAE syndromes [3, 39, 79]. While rarer, a feature of EMBE is the paucity of systemic complications, in clear contrast with HSES. This distinction raises suspicion that observed cases of IAE may be manifestations of a common pathophysiological process in varying degrees of severity.
Meanwhile, a common feature between reported cases of EMBE is the clinically-correlated lack of radiological abnormalities with the exception of diffuse brain swelling. Outcomes are polarized between full neurological recovery and fatal cerebral herniation [39, 79]. Mainstays of treatment are supportive with careful management of intracranial pressure. Sakurai et al [79]. reported a case with complete neurological recovery, following treatment with oseltamivir, methylprednisolone, intravenous immunoglobulins, induced hypothermia, and intravenous anti-thrombin III.
Hemorrhagic Shock and Encephalopathy Syndrome
HSES is a clinically defined syndrome initially described by Levin et al. in 1983. It is a severe disease that seems to exclusively affect the pediatric population. Defining clinical features include shock, coma or seizures, diarrhea, disseminated intravascular coagulopathy (DIC), drop in hemoglobin and platelet, elevated liver enzymes, renal dysfunction, acidosis, and negative blood and cerebrospinal fluid cultures. “Definite HSE” is defined by satisfaction of all nine criteria, and “probable HSES” requires the presence of at least seven of the above with two unknown, or satisfaction of eight criteria with one unmet criterion [6]. Patients typically present with an acute history of watery or bloody diarrhea, followed by shock, seizures, and altered mental status. Disease-defining systemic complications ensue. A biphasic course of illness is occasionally noted, with transient improvement in neurological status at 12–24 h, followed by subsequent deterioration and similarly poor outcomes [26].
EEG shows early focal or multifocal spikes with possible decrease in signal amplitude and diffuse slowing as early as within the first 72 h, although a minority of patients may still have normal EEG findings. Subsequent slowing reflective of encephalopathic state is common [6]. Diffuse cerebral edema with ventricular effacement may be apparent on CT studies within the first 3 days [57]. MRI may further delineate areas of prominent hypointensity on T2-weighted images due to hemosiderin deposition, correlating with pathologic findings of hemorrhagic necrosis with liquefaction involving the cerebral cortex, basal ganglia, and hemispheric white matter [26].
As a clinical syndrome solely defined by specific organ systems affected, it is conceivable that HSES is also associated with other pathogens though many known cases have no identified microbiological etiology [6, 26, 28]. Independently of pathogen, HSES has high mortality and poor clinical functional outcome on survival. Acute-phase mortality is estimated at 60 %, and 30 % of survivors have neurological sequelae [6, 26].
Acute Necrotizing Encephalopathy
ANE is the most commonly reported neurological complication of influenza A infection, affecting both pediatric and adult populations. The characteristic clinical presentation includes fever and prodrome of upper respiratory tract infection, followed by rapid and severe decline in level of consciousness [56]. Seizures may be present at onset, and are reported to be often refractory to anti-seizure medications. Neurological symptoms are noted on average within 1–5 days of systemic symptom onset; progression to death may occur in as little as 1 day after onset of clinically apparent infection, in certain fulminant cases reported [44, 56]. A major challenge in the management of a patient with suspected ANE is the initial ascertainment of the diagnosis. Differential diagnoses to consider include metabolic or toxic encephalopathy, infectious, vascular, or neoplastic pathologies. Certain uncommon viral illnesses may present similarly, including HHV-6, acute measles encephalitis, West Nile, Japanese encephalitis, Murray Valley, and Eastern equine encephalitis viruses, and rabies [53]. It is equally important to note that ANE is a clinical syndrome associated with, but not defining, an influenza infection [56]. However, it remains one of the most severe influenza-associated neurological complication.
Cerebrospinal fluid in ANE shows mild pleocytosis, but often is of limited diagnostic value. MRI findings suggestive of ANE include symmetric, diffusion-restricting lesions in both thalami, rostral midbrain tegmentum, putamina, periventricular white matter and cerebellar hemispheres [56]. These area of abnormal MRI signals correspond to macroscopically noted hemorrhagic necrosis on autopsy. In limited angiographic data, decreased flow without stenosis or thrombosis has been noted in the arterial branches supplying these areas, notably the thalamoperforating, thalamogeniculate and superior cerebellar arteries, and in the deep draining venous system, notably the internal and great cerebral veins [56]. Macroscopic pathological examination shows severe edema and necrosis in the gray matter of the thalamic, tegmental and cerebellar dentate nucleus. Similar edema is diffusely noted in the cerebral and cerebellar white matter. Microscopic findings show expansion of perivascular space with a small number of extravasated leukocytes and otherwise no signs of inflammatory infiltrates or vascular necrosis [56, 57, 64]. Therefore, ANE is not an encephalitis but an acute encephalopathy associated with clear pathologic changes.
Early use of corticosteroids has been suggested to confer improved survival and cognitive outcomes specifically for ANE patients without brainstem lesions, likely through modification of the overwhelming inflammatory response responsible for blood–brain barrier disruption [69]. It is unclear whether the absence of benefit from corticosteroids in patients with brainstem lesions is related to an alternate pathophysiology or the reflection of an already aggressive disease course, as many of these patients are at risk for sudden, neurogenic cardiopulmonary collapse [69]. Alternative immunosuppressive therapies such as Cyclosporine have also been proposed [69].
Despite early steroid therapy, Okumura et al [69]. reported a mortality rate of approximately 30 % in a case series of influenza-associated ANE, with another 33 % of patients left with severe cognitive and motor impairment. While the primary cause of mortality in patients with ANE is, in most instances, from cardiorespiratory failure, brain death has been reported in a number of case reports [54].
Acute Encephalopathy with Seizures and Late Restricted Diffusion
AESD likely represents a syndrome described and known by various similar titles, such as “acute encephalopathy with febrile convulsive status epilepticus”, “acute encephalopathy with prolonged febrile seizures and late reduced diffusion”, or “encephalopathy with biphasic clinical course” [57]. AESD is a predominantly pediatric diagnosis, with a clear and disease-defining biphasic course. It typically begins with febrile generalized tonic–clonic seizures and post-ictal decreased level of consciousness over the first 24 h with subsequent normalization in neurological status. It may be indistinguishable from other febrile convulsive status epilepticus in this initial phase. Over 4–6 days following onset of symptoms, clusters of secondarily generalized seizures recur, with or without fever [86, 88]. Interictal normalization in mental status only occurs in a minority of patients at this stage. With resolution of seizures on days 4–18, patients may manifest abnormal neurological signs, such as choreoathetosis, hand writhing and other stereotypical involuntary movements, aphasia, motor apraxia, cognitive and behavioral abnormalities, or hemiparesis [57].
Investigations reveal normal CSF cell and protein contents. Serum transaminases may be normal or mildly elevated. Both hyperglycemia and hypoglycemia may be noted. Importantly, in the presence of hypoglycemia and diffuse intravascular coagulopathy, Reye’s syndrome or HSES must be considered as differential diagnoses portending poorer outcomes. Unlike in complex febrile seizures, serum interleukin-6 (IL-6) levels are mild-to-moderately elevated [57]. While initially normal, MRI performed after 2 days can show hyperintensity on T2-weighted imaging or diffusion-restriction in the frontal or fronto-parietal U-fibers and cortex. A “bright-tree” appearance on DWI is characteristic, with frequent sparing of the peri-Rolandic region [57, 83, 88]. Corresponding affected cortices are noted to be hyperperfused on single photon-emission computed tomography [57, 88]. Resolution begins within one to 4 weeks, as clinically milder cases may show radiological normalization and more severe cases progress to atrophy and hypoperfusion of the cortical areas initially affected [57].
Based on clinical symptoms and signs, Mizuguchi et al [57, 94]. proposed a further subdivision of AESD into a predominantly infantile variant with evidence of selective frontal pathology and another affecting an entire hemisphere. Yamanouchi and Mizuguchi described a pediatric subset of patients with features of AESD meeting four characteristics as AIEF [94]. The proposed diagnostic criteria are: (1) acute encephalopathy of infancy or early childhood following a viral illness; (2) symptoms and signs of frontal lobe dysfunction; (3) radiological evidence of selective frontal cortical edema and hyperperfusion followed by atrophy and hypoperfusion; and (4) the absence of evidence of alternate inflammatory or metabolic disorders [95]. Likewise, HHS may be considered a variant of AESD with post-ictal hemiparesis, cognitive decline and seizure disorder with similar radiographic findings of acute cortical edema and hyperperfusion, and subsequent atrophy [57]. While HHS is defined as a pediatric condition, Chen et al [11]. reported a case bearing remarkable similarities in clinical course, findings and outcomes in a 40-year-old man with IAE secondary to novel H1N1 influenza A virus.
Outcomes associated with AESD range from normal or mild cognitive impairment to severe mental retardation, paralysis, or epilepsy. Mortality rate has been reported at less than 5 % [57].
The Novel H1N1 Influenza Pandemic of 2009–2010
In April 2009, outbreak of a novel strain of influenza A (H1N1) virus began in Mexico, followed by cases reported in the USA and across the world. In a review by the Writing Committee of the WHO Consultation on Clinical Aspects of Pandemic (H1N1) 2009 Influenza, by May 2010, the novel H1N1 pandemic accounted for six million reports cases, 270,000 hospitalizations, and over 12,000 deaths [62]. It is believed that the true incidence of novel H1N1-related infections in the pediatric population is underestimated, and likely better approximated at 33 %, based on serological evidence [8, 62]. Clinically significant illness in young adults occurred more frequently than with seasonal influenza, with a relative sparing of the population over 60 years of age, postulated to be due to cross-immunity conferred by the 1957 H1N1 epidemic [8]. Despite a seemingly low case fatality rate of 0.048 % in the USA and 0.026 % in the UK, 90 % of reported deaths occurred in individuals less 65 years old, representing a remarkable contrast from seasonal influenza [56, 72, 96].
In July 2009, Evans et al. [62] published the first report of H1N1-associated neurological complications, consisting of four pediatric cases of seizures and encephalopathy with onset within 5 days of proven systemic influenza illness. Numerous case reports followed, indicating the novel H1N1’s association with a wide range of neurological complications. The Japanese Society of Intensive Care Medicine and Respiratory Care Medicine’s national registries recorded that 23 % of children admitted to the Critical Care Unit had various neurological symptoms [89]. Syndromes reported in different world regions were largely similar to prior influenza pandemics and seasonal epidemics in spectrum and severity: influenza-associated encephalopathy, seizures, Guillain-Barré syndrome, and rhabdomyolysis [8, 12, 41, 53, 62, 71, 72, 96]. A case of ischemic stroke in a 9-month-old infant has also been reported [30]. Of particular interest is an observed sudden 17-fold increase in pediatric narcolepsy in 2010, following the pandemic. The etiology of this surge is unclear, but postulated to be secondary to both genetic and environmental factors [73]. Reye’s syndrome’s remarkable absence in the literature pertaining to the 2009 H1N1 pandemic may be attributed to its well-recognized association with acetylsalicylic acid. Meanwhile, atypical presentations have been observed. For instance, Fugate et al. [23] reported a case of acute hemorrhagic leukoencephalitis in a 40-year-old, otherwise healthy man, after prolonged cardiorespiratory support following H1N1 infection, resulting in severe disability.
First-line treatment options for novel H1N1 are neuraminidase inhibitors, most commonly oseltamivir [62]. As oseltamivir is noted to have limited cerebrospinal fluid penetration, it likely has limited activity for infections of the central nervous system [80]. However, it reduces duration of hospitalization, severity of systemic disease, and consequent ICU admission or mortality [9]. As most influenza-associated neurological complications are non-fatal, but often co-occurring with severe cardiopulmonary disease or mortality, treatment is recommended [62]. However, oseltamivir-resistant strains exist, frequently through a neuraminidase-dependent mechanism. The common point mutation of the viral neuraminidase gene causing oseltamivir resistance does not simultaneously confer cross-resistance to zanamivir, which may be used as a second-line agent [91]. A fatal case of multidrug-resistant H1N1 virus has also been reported during the 2009 pandemic, with marked increase in the 50 % inhibitory concentration to oseltamivir, zanamivir, and the new neuraminidase inhibitor peramivir, leaving the clinician with few treatment options [91].
Influenza Vaccine-Associated Complications
Inflammatory peripheral and central demyelinating diseases, such as Guillain-Barré syndrome (GBS) and acute disseminated encephalomyelitis (ADEM), are well-recognized complications of seasonal influenza vaccination, occurring in both pediatric and adult populations [35, 43, 61]. Neurological complications of lesser severity have also been reported. Severe vasculitic mononeuritis multiplex, Bell’s palsy, optic perineuritis, and giant cell arteritis have been noted to occur in close temporal association with influenza vaccine [19, 34, 74, 92].
Safety of seasonal influenza vaccine has also been studied for various neurological and autoimmune diseases. No increase in the risk of myasthenia gravis, multiple sclerosis or systemic autoimmune disease exacerbations was identified following influenza vaccination [5, 7, 50, 99]. Given the likelihood of poorer prognosis if affected by respiratory complications of influenza, the vaccine likely confers protection in these patients often subject to chronic immunosuppressive therapies. Influenza vaccination has also been correlated with decreased relative risks of embolic stroke [65, 84].
Safety Data of the H1N1 Vaccine
Concerns regarding H1N1 vaccines emanated from the 1976–77 season data, when a surge in the incidence of GBS led to premature cessation of the vaccination campaign in the USA [18, 71, 78]. GBS affected an estimated rate of 7.2 per million vaccinated individuals, over the first 6 weeks after receiving the A/NJ/1976 vaccine, versus a risk of 0.79 cases per million unvaccinated people, yielding an attributable risk of 8.8 per million recipients [18]. Subsequent in vivo studies have demonstrated the A/NJ/1976 vaccine’s ability to induce antibodies to epitopes mimicking the monosialoganglioside GM1, well-known to be involved in some cases of GBS. However, numerous vaccines used in past campaigns had similar laboratory behavior, yet different clinical and epidemiological responses [17, 18]. The etiology of the observed rise in GBS following the 1976 H1N1 vaccination campaign remains unclear. [18].
Over the past decades, numerous large-scale databases have been established for surveillance of neurological adverse effects related to the vaccine. These include the American Vaccine Adverse Event Reporting System and the Vaccine Surveillance Database, a population-based computerized encounter code database. They allow rapid analyses of the number of individuals receiving vaccines and subsequent complications [10, 15, 56, 78]. However, many adverse effects are reported weeks after vaccination, which poses a challenge in establishing a certain causal link between two clinical events [10, 18, 42, 52, 61]. Moreover, most entries are preliminary diagnoses, inevitably including some erroneous data [55]. Para- and post-infectious GBS have also been increasingly reported as infectious complications of influenza A virus, further complicating the vaccine’s risk–benefit analysis [10, 52]. The 2009 H1N1 vaccination campaign reported an attributable risk of 0.8 cases per million individuals for developing GBS, similar to that of seasonal influenza vaccines, and ten-fold less than that recorded in 1976, with an overall risk profile identical to seasonal influenza vaccines [35, 75, 78]. No safety concerns were identified in vaccinated pregnant patients after the 2009 pandemic [58].
Pathophysiology of Influenza-Associated Complications
Direct tissue invasion has long been the suspected pathophysiology behind influenza virus’s neurological complications, and viral genomes have been demonstrated on sampling of cerebrospinal fluid (CSF), in a small number of patients. However, subsequent studies failed to replicate similar results [24, 40, 45]. Shieh et al. reviewed pathology results of 100 fatal cases of the 2009 H1N1 pandemic, and did not isolate the influenza A virus from any post-mortem cerebral tissues [82]. This observation is in contrast to a more frequently observed myotropism in influenza-associated myositis (IAM), a peripheral nervous system complication of influenza disease. As viral particles have more often been identified on tissue sampling, it is speculated that IAM can indeed be a consequence of direct infection, unlike IAE [1, 12].
Given associated manifestations often associated with cytokine dysregulation, including hemophagocytosis in 50 % of the subjects, several authors suggested hypercytokinemia to be a most likely pathophysiological etiology of clinically observed H1N1-associated encephalopathy [82, 93].
Pathophysiology of IAE
Increasing evidence suggest hypercytokinemia with secondary blood–brain barrier disruption and neuronal apoptosis to be responsible for the observed diffuse cerebral dysfunction and pathologies [36, 46, 51, 60, 66]. Furthermore, in one autopsy series, hemophagocytosis was seen on autopsy of over 50 % of patients with influenza-related deaths, with or without encephalopathy. As hemophagocytosis is believed to be a cytokine-driven response, the observation supports the pathophysiological role of a possible overwhelming and dysregulated inflammatory state [14, 93]. Oxidative stress and free radical gases also appear to participate in the pathogenesis of IAE [48, 49].
Fluctuations in inflammatory markers are therefore important in elucidating IAE’s pathogenesis and studied as potentially clinically useful markers of disease severity. Candidate serum markers include interleukin-6 (IL-6), tumor necrosis factor alpha (TNF-α), soluble TNF receptor-1, interleukin-8 (IL-8), interleukin-1β (IL-1β), interleukin-10 (IL-10), CD40-ligand (CD40L), matrix metalloproteinase-9 (MMP-9), and toll-like receptor 3 [29, 31, 36–38, 46, 66] (Table 2). Cerebrospinal fluid nitrite and nitrate (NOx), reactive oxygen metabolites, and cytochrome c levels have also been considered [31, 32, 48, 49].
Interleukin-6 (IL-6) is known to confer neuroprotection in physiological states, but serum IL-6 has been noted to be disproportionately elevated in IAE, correlating with unfavorable outcomes, including severe disability and death [2, 31, 61]. A surge in serum IL-6 can be noted over 12 h prior to the onset of encephalopathy. The pathophysiological significance of this rise is unclear. It was proposed that this observation may be interpreted as evidence of immune dysregulation in IAE, or that IL-6 overexpression subsequently induces or contributes to NMDA-mediated neurotoxicity, in pathological states [2]. Independently of its pathophysiologic role, IL-6 level seemed to correlate reliably with severity of disease.
Elevated serum TNF-α has similarly been demonstrated [27, 49]. While TNF-α is known to alter blood–brain barrier permeability, it may equally play a role in NMDA-mediated neurotoxicity [2, 36]. Serum TNF-α levels over 10 pg/mL at presentation, and especially if over 50 pg/mL, were reported to be associated with poor prognosis [32]. Serum soluble TNF receptor binds TNF-α, and its upregulation has been invoked as the most sensitive measure in reflection of TNF-α elevation [36]. Other proinflammatory cytokines, such as IL-1β and IL-8, were observed to be elevated, although their significance in IAE remains unclear [46]. Elevated serum CD40L is similarly noted in patients with poor outcome in IAE, suggesting platelet activation in the early inflammatory response [32]. Meanwhile, overproduction of the anti-inflammatory cytokine IL-10 is also noted in IAE. Its role as part of the counter-regulatory response has been correlated with mortality in sepsis, through possible dysregulation and consequent immunological anergy. A similar mechanism may be at play in IAE [46].
Serum MMP-9 and tissue inhibitors of metalloproteinases (TIMP), its regulatory counterparts, play key roles in mediating blood–brain barrier function, and are known to be involved in post-thrombolytic cerebral hemorrhage and post-influenza embolic strokes [37, 60]. Alterations in their levels are therefore no surprises in IAE, consistent with the frequent observation of focal or diffuse cerebral edema. Ichiyama et al. reported IAE patients’ serum MMP-9 levels and MMP-9/TIMP-1 ratios to be in excess of that observed in other viral infections such as Ebstein-Barr virus (EBV) and respiratory syncitial virus [37].
Serum cytochrome c also show a rise in the early phase of disease and may be the best marker of poor outcome in IAE [31, 32, 66]. It is estimated to have a sensitivity and specificity estimated at 93 and 100 %, respectively, for acute encephalopathy in children [31]. Different cutoffs were reported at 1000 pg/mL or 45 ng/mL [31, 32]. CSF cytochrome c has been studied as a marker of apoptosis in patients with IAE: increase in neuronal cytoplasmic levels activates caspase-mediated neuronal apoptosis [32]. While high serum cytochrome c on admission is almost uniformly associated with fatal outcome, progressive rise in the level of CSF cytochrome c is closely correlated with high morbidity. As cytochrome c is not transported across the blood–brain barrier, its rise in CSF is suggestive of increased intraneuronal expression: it likely plays a direct role in initiating apoptosis in severe IAE. Its levels correlate closely with subsequent degrees of cerebral atrophy [32, 66].
Role of Host Immunity
Hypercytokinemia implies that the infected host plays a role in the pathogenesis of influenza-associated neurological complications. This theory is supported by two observations: ethnic predisposition for influenza-associated neurological complications, and known familial cases of recurrent influenza-associated ANE [27].
Japan and Taiwan have reported a disproportionate number of influenza-associated complications over the past decade. The epidemiological impact of these conditions have led to a well-established surveillance system in Japan. The etiology of this preponderance has been questioned, but remains unclear. However, influenza-associated neurological complications are not exclusive to East Asian populations, and both sporadic and pandemic cases with identical natural history have been diagnosed in Caucasian patients, and reported in the medical literature from various continents [3, 70, 77].
Genetic predisposition to the development of ANE has been studied following recurrent presentations, affecting both mother and child. Gika et al [27]. identified mutations in Ran-binding protein 2 (RANBP2), a nuclear pore protein component on chromosome 2q12.1-q13, as susceptibility alleles for recurrent or familial ANE, termed ANE1. Its inheritance follows an autosomal dominant pattern with a penetrance of 40 %, and its clinical behavior differs from the sporadic variant. Many cases follow a fulminant course, and it is believed that ANE1 favors involvement of the claustrum, limbic system, and temporal lobes. The authors underline that the RANBP2 protein also localizes to microchondria and microtubules, hence further data is required to define ANE1 as a nuclear pore disease, despite its implications in the pathophysiology of infection as well [27]. Mitochondrial carnitine palmitoyltransferase II (CPT II) is suspected to be one of such proteins, as certain compound variants are considered phenotypically normal, but show significantly decreased metabolic activity during high fever [97]. Consequent impairments in mitochondrial fuel utilization in susceptible individuals may contribute to the development of cerebral edema in IAE [49].
Managing Influenza-Associated Neurological Complications
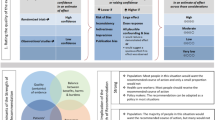
As per general clinical management principles, ensuring patient stability should be the first priority. Thereon, ascertainment of the diagnosis is key in appropriately managing influenza-associated neurological complications (IANC). (Figures 1, 2) Key elements on history are acute alterations in consciousness following recent influenza disease, exposure or vaccination, with a clear timeline of symptom progression, and a detailed review of systems. Physical examination should be focused on determining the severity of the alteration in mental status and signs of focal neurological disease, and broadened to identify evidence of systemic complications, including signs of thrombocytopenia and coagulopathy.

Initial management and investigations in suspected influenza-associated acute encephalopathy. It is most important to recognize that a high index of suspicion is primal in ascertaining the diagnosis. MRI—magnetic resonance imaging; CSF—cerebrospinal fluid; EEG—electroencephalogram; IAE—influenza-associated encephalopathy; CBC—complete blood count; PCR—polymerase chain reaction; HSV—herpes simplex virus; VZV—varicella zoster virus; EBV—Ebstein-Barr virus; HHV-6—human herpesvirus-6

Diagnosis and management of major variants of influenza-associated encephalopathy. ALOC—alteration in level of consciousness; DIC—diffuse intravascular coagulopathy; CSF—cerebrospinal fluid; MRI—magnetic resonance imaging; EEG—electroencephalogram; MERS—mild encephalopathy with reversible splenial lesion; AESD—acute encephalopathy with seizures and late restricted diffusion; HSES—hemorrhagic shock and encephalopathy syndrome; ANES—acute necrotizing encephalopathy syndrome; IL-6—interleukin-6
Initial laboratory investigations should include complete blood count, electrolyte levels, coagulation profile, renal and hepatic function tests, liver enzyme levels, creatinine phosphokinase level, bacterial blood cultures, nasopharyngeal swab and lumbar puncture. CSF chemistry profile, cell count, and microbiological studies including viral PCR for influenza, adenovirus and enteroviruses, bacterial, and fungal cultures. Rarer viral infections should be ruled out based on the patient’s history and exposure. A brain magnetic resonance study is recommended, including at least basic T1- and T2-weighted sequences and diffusion-weighted images. A screening brain CT scan should be performed when the MRI is not readily available. Electroencephalograms findings in IANC are generally non-specific and of low diagnostic yield, and would only be recommended in management of suspected non-convulsive seizures. Following the aforementioned diagnostic investigations, if suspicion of IANC is high, serum IL-6 and cytochrome c levels are recommended where available, mostly for prognostication purposes.
In managing influenza-associated complications of the central nervous system (CNS), it is important to recognize that mortality is most frequently a consequence of systemic illness, while CNS disease portends the highest risk of disability on survival. Symptomatic treatment of CNS complications, management of associated cardiopulmonary, metabolic and hematological complications, and targeted anti-inflammatory therapies and anti-viral therapies are recommended. Close monitoring for expected complications through serial laboratory studies and a low threshold for repeat radiological investigations are equally warranted.
Targeted anti-inflammatory therapies are most likely to impact or alter the clinical course. Initiation of corticosteroid therapy within 24 h of admission has been demonstrated to confer survival benefit in patients with ANE and without brainstem disease and has been recommended by the Research Organization for Influenza Encephalopathy Researchers of Japan since 1999 [69]. Okuruma et al. have demonstrated no significant difference in outcome between the use of pulsed methylprednisolone at a dose of 30 mg/kg/day for 3 days or intravenous dexamethasone at a dose of 0.6 mg/kg/day in four divided dose over 2–4 days. Despite a limited number of patient studied, similar outcome was also noted with intravenous immunoglobulin at a dose of 1–2 mg/kg [68] plasma exchange directed toward reduction of serum IL-6 levels have been attempted, for a total of two or three treatments administered every other day, with reported good outcomes [47]. Supportive care for CNS complications should also include intracranial pressure monitoring and management, where osmotic agents are usually sufficient. Mild hypothermia with target core temperature of 34 °C for 3 days with subsequent rewarming at a rate of 1 °C per day has been proposed [98].
Conclusion
Influenza-associated neurological complications have become increasingly recognized over the past century. While the incidence remains low in most parts of the world and is mostly accounted for by the pediatric population, these clinical entities are not restricted by ethnicity, geographical area, or patient age, especially during pandemics. Defining recurrent syndromes instead of individual neurological symptoms provides the benefit of timely diagnosis of a potentially aggressive, fatal or disabling disease, especially when early treatment may decrease morbidity and mortality. Where clinical presentation differs from recognized disease patterns, the physician should seek authorization with substitute decision makers for biopsy, or autopsy in fatal cases, to ascertain clinical, microbial, and pathological correlates. Further data is necessary to expand our understanding and solidify the international experience with influenza-associated neurological complications, and may even open the gateway to better understanding of acute encephalopathies secondary to dysregulated systemic inflammatory response, of both infectious and non-infectious etiologies.
References
Agyeman P, Duppenthaler A, Heininger U, Aebi C. Influenza-associated myositis in children. Infection. 2004;32(4):199–203.
Aiba H, Mochizuki M, Kimura M, Hojo H. Predictive value of serum interleukin-6 level in influenza virus-associated encephalopathy. Neurology. 2001;57(2):295–9.
Akins PT, Belko J, Uyeki TM, Axelrod Y, Lee KK, Silverthorn J. H1N1 encephalitis with malignant edema and review of neurologic complications from influenza. Neurocrit Care. 2010;13(3):396–406.
Amin R, Ford-Jones E, Richardson SE, MacGregor D, Tellier R, Heurter H, Fearon M, Bitnun A. Acute childhood encephalitis and encephalopathy associated with influenza: a prospective 11 year review. Pediatr Infect Dis J. 2008;27(5):390–5.
Auriel E, Gadoth A, Regev K, et al. Seasonal and H1N1v influenza vaccines in MS: safety and compliance. J Neurol Sci. 2012;314(1–2):102–3.
Bacon CJ, Hall SM. Haemorrhagic shock encephalopathy syndrome in the British Isles. Arch Dis Child. 1992;67(8):985–93.
Bardage C, Persson I, Ortgvist A, et al. Neurological and autoimmune disorders after vaccination against pandemic influenza A (H1N1) with a monovalent adjuvanted vaccine: population based cohort study in Stockholm. Sweden BMJ. 2011;343:d5956.
Bautista E, Chotpitayasunondh T, Gao Z, Harper SA, Shaw M, Uyeki TM, Zaki SR, Hayden FG, Hui DS, Kettner JD, et al. Clinical aspects of pandemic 2009 influenza A (H1N1) virus infection. N Engl J Med. 2010;362(18):1708–19.
Berger JR, Houff SA. Neurological infections: the year of PML and influenza. Lancet Neurol. 2010;9(1):14–7.
Black S, Eskola J, Siegrist CA, Halsey N, Macdonald N, Law B, Miller E, Andrews N, Stowe J, Salmon D, et al. Importance of background rates of disease in assessment of vaccine safety during mass immunisation with pandemic H1N1 influenza vaccines. Lancet. 2009;374(9707):2115–22.
Chen YC, Lo CP, Chang TP. Novel influenza A (H1N1)-associated encephalopathy/encephalitis with severe neurological sequelae and unique image features–a case report. J Neurol Sci. 2010;298(1–2):110–3.
Davis LE. Neurologic and muscular complications of the 2009 influenza A (H1N1) pandemic. Curr Neurol Neurosci Rep. 2010;10(6):476–83.
de Jong MD, Bach VC, Phan TQ, Vo MH, Tran TT, Nguyen BH, Beld M, Le TP, Truong HK, Nguyen VV, et al. Fatal avian influenza A (H5N1) in a child presenting with diarrhea followed by coma. N Engl J Med. 2005;352(7):686–91.
de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, Hoang DM, Chau NV, Khanh TH, Dong VC, et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med. 2006;12(10):1203–7.
Destefano F, Tokars J. H1N1 vaccine safety monitoring: beyond background rates. Lancet. 2010;375(9721):1146–7.
Drago L, Attia MW, DePiero A, Bean C. Influenza A-associated meningoencephalitis. Pediatr Emerg Care. 2005;21(7):437–9.
Eisen DP, McBryde ES. Avoiding Guillan-Barre Syndrome following swine origin pandemic H1N1 2009 influenza vaccination. J Infect Dis. 2009;200(10):1627–8.
Evans D, Cauchemez S, Hayden FG. “Prepandemic” immunization for novel influenza viruses, “swine flu” vaccine, Guillain-Barre syndrome, and the detection of rare severe adverse events. J Infect Dis. 2009;200(3):321–8.
Finsterer J, Artner C, Kladosek A, Kalchmayr R, Redtenbacher S. Cavernous sinus syndrome due to vaccination-induced giant cell arteritis. Arch Intern Med. 2001;161(7):1008–9.
Fluss J, Ferey S, Menache-Starobinski C, Delavelle J, Van Bogaert P, Vargas MI. Mild influenza-associated encephalopathy/encephalitis with a reversible splenial lesion in a Caucasian child with additional cerebellar features. Eur J Paediatr Neurol. 2010;14(1):97–100.
Foley PB. Encephalitis lethargica and the influenza virus. II. The influenza pandemic of 1918/19 and encephalitis lethargica: epidemiology and symptoms. J Neural Transm. 2009;116(10):1295–308.
Foley PB. Encephalitis lethargica and the influenza virus. III. The influenza pandemic of 1918/19 and encephalitis lethargica: neuropathology and discussion. J Neural Transm. 2009;116(10):1309–21.
Fugate JE, Lam EM, Rabinstein AA, Wijdicks EF. Acute hemorrhagic leukoencephalitis and hypoxic brain injury associated with H1N1 influenza. Arch Neurol. 2010;67(6):756–8.
Fujimoto S, Kobayashi M, Uemura O, Iwasa M, Ando T, Katoh T, Nakamura C, Maki N, Togari H, Wada Y. PCR on cerebrospinal fluid to show influenza-associated acute encephalopathy or encephalitis. Lancet. 1998;352(9131):873–5.
Ganapathy S, Ey EH, Wolfson BJ, Khan N. Transient isolated lesion of the splenium associated with clinically mild influenza encephalitis. Pediatr Radiol. 2008;38(11):1243–5.
Gefen R, Eshel G, Abu-Kishk I, Lahat E, Youngster I, Rosenbloom E, Kozer E. Hemorrhagic shock and encephalopathy syndrome: clinical course and neurological outcome. J Child Neurol. 2008;23(5):589–92.
Gika AD, Rich P, Gupta S, Neilson DE, Clarke A. Recurrent acute necrotizing encephalopathy following influenza A in a genetically predisposed family. Dev Med Child Neurol. 2010;52(1):99–102.
Gooskens J, Kuiken T, Claas EC, Harinck HI, Thijssen JC, Baelde HJ, Kroes AC. Severe influenza resembling hemorrhagic shock and encephalopathy syndrome. J Clin Virol. 2007;39(2):136–40.
Hidaka F, Matsuo S, Muta T, Takeshige K, Mizukami T, Nunoi H. A missense mutation of the Toll-like receptor 3 gene in a patient with influenza-associated encephalopathy. Clin Immunol. 2006;119(2):188–94.
Honorat R, Tison C, Sevely A, et al. Influenza A(H1N1)-associated ischemic stroke in a 9-month-old child. Pediatr Emerg Care. 2012;28(4):368–9.
Hosoya M, Kawasaki Y, Katayose M, Sakuma H, Watanabe M, Igarashi E, Aoyama M, Nunoi H, Suzuki H. Prognostic predictive values of serum cytochrome c, cytokines, and other laboratory measurements in acute encephalopathy with multiple organ failure. Arch Dis Child. 2006;91(6):469–72.
Hosoya M, Nunoi H, Aoyama M, Kawasaki Y, Suzuki H. Cytochrome c and tumor necrosis factor-alpha values in serum and cerebrospinal fluid of patients with influenza-associated encephalopathy. Pediatr Infect Dis J. 2005;24(5):467–70.
Huang YC, Lin TY, Wu SL, Tsao KC. Influenza A-associated central nervous system dysfunction in children presenting as transient visual hallucination. Pediatr Infect Dis J. 2003;22(4):366–8.
Hull JH, Mead SH, Foster OJ, Modarres-Sadeghi H. Severe vasculitic neuropathy following influenza vaccination. J Neurol Neurosurg Psychiatry. 2004;75(10):1507–8.
Huynh W, Cordato DJ, Kehdi E, Masters LT, Dedousis C. Post-vaccination encephalomyelitis: literature review and illustrative case. J Clin Neurosci. 2008;15(12):1315–22.
Ichiyama T, Isumi H, Ozawa H, Matsubara T, Morishima T, Furukawa S. Cerebrospinal Fluid and Serum Levels of Cytokines and Soluble Tumor Necrosis Factor Receptor in Influenza Virus-associated Encephalopathy. Scand J Infect Dis. 2003;35(1):59–61.
Ichiyama T, Morishima T, Kajimoto M, Matsushige T, Matsubara T, Furukawa S. Matrix metalloproteinase-9 and tissue inhibitors of metalloproteinases 1 in influenza-associated encephalopathy. Pediatr Infect Dis J. 2007;26(6):542–4.
Ichiyama T, Morishima T, Suenaga N, Kajimoto M, Matsubara T, Furukawa S. Analysis of serum soluble CD40 ligand in patients with influenza virus-associated encephalopathy. J Neurol Sci. 2005;239(1):53–7.
Ishigami A, Kubo S, Ikematsu K, Kitamura O, Tokunaga I, Gotohda T, Nakasono I. An adult autopsy case of acute encephalopathy associated with influenza A virus. Leg Med (Tokyo). 2004;6(4):252–5.
Ito Y, Ichiyama T, Kimura H, Shibata M, Ishiwada N, Kuroki H, Furukawa S, Morishima T. Detection of influenza virus RNA by reverse transcription-PCR and proinflammatory cytokines in influenza-virus-associated encephalopathy. J Med Virol. 1999;58(4):420–5.
Iwata A, Matsubara K, Nigami H, Kamimura K, Fukaya T. Reversible splenial lesion associated with novel influenza A (H1N1) viral infection. Pediatr Neurol. 2010;42(6):447–50.
Juurlink DN, Stukel TA, Kwong J, Kopp A, McGeer A, Upshur RE, Manuel DG, Moineddin R, Wilson K. Guillain-Barre syndrome after influenza vaccination in adults: a population-based study. Arch Intern Med. 2006;166(20):2217–21.
Kao CD, Chen JT, Lin KP, Shan DE, Wu ZA, Liao KK. Guillain-Barre syndrome coexisting with pericarditis or nephrotic syndrome after influenza vaccination. Clin Neurol Neurosurg. 2004;106(2):136–8.
Kasai T, Togashi T, Morishima T. Encephalopathy associated with influenza epidemics. Lancet. 2000;355(9214):1558–9.
Kawada J, Kimura H, Hara S, Ito Y, Kawashima H, Okuno T, Morishima T. Absence of associations between influenza-associated encephalopathy and human herpesvirus 6 or human herpesvirus 7. Pediatr Infect Dis J. 2003;22(2):115–9.
Kawada J, Kimura H, Ito Y, Hara S, Iriyama M, Yoshikawa T, Morishima T. Systemic cytokine responses in patients with influenza-associated encephalopathy. J Infect Dis. 2003;188(5):690–8.
Kawashima H, Togashi T, Yamanaka G, Nakajima M, Nagai M, Aritaki K, Kashiwagi Y, Takekuma K, Hoshika A. Efficacy of plasma exchange and methylprednisolone pulse therapy on influenza-associated encephalopathy. J Infect. 2005;51(2):E53–6.
Kawashima H, Watanabe Y, Ichiyama T, Mizuguchi M, Yamada N, Kashiwagi Y, Takekuma K, Hoshika A, Mori T. High concentration of serum nitrite/nitrate obtained from patients with influenza-associated encephalopathy. Pediatr Int. 2002;44(6):705–7.
Kawashima H, Watanabe Y, Morishima T, Togashi T, Yamada N, Kashiwagi Y, Takekuma K, Hoshika A, Mori T. NOx (nitrite/nitrate) in cerebral spinal fluids obtained from patients with influenza-associated encephalopathy. Neuropediatrics. 2003;34(3):137–40.
Kostianovsky A, Charles P, Alves JF, et al. Immunogenicity and safety of seasonal and 2009 pandemic A/H1N1 influenza vaccines for patients with autoimmune diseases: a prospective, monocentre trial on 199 patients. Clin Exp Rheumatol. 2012;30(1 Suppl 70):S83–9.
Lee N. Acute Encephalopathy Associated with Influenza A Infection in Adults. Emerging Infectious Diseases 2010;16(1).
Lehmann HC, Hartung HP, Kieseier BC, Hughes RA. Guillain-Barre syndrome after exposure to influenza virus. Lancet Infect Dis. 2010;10(9):643–51.
Lyon JB, Remigio C, Milligan T, Deline C. Acute necrotizing encephalopathy in a child with H1N1 influenza infection. Pediatr Radiol. 2010;40(2):200–5.
Martin A, Reade EP. Acute necrotizing encephalopathy progressing to brain death in a pediatric patient with novel influenza A (H1N1) infection. Clin Infect Dis. 2010;50(8):e50–2.
Maurizi CP. Influenza caused epidemic encephalitis (encephalitis lethargica): the circumstantial evidence and a challenge to the nonbelievers. Med Hypotheses. 2010;74(5):798–801.
Mizuguchi M. Acute necrotizing encephalopathy of childhood: a novel form of acute encephalopathy prevalent in Japan and Taiwan. Brain Dev. 1997;19(2):81–92.
Mizuguchi M, Yamanouchi H, Ichiyama T, Shiomi M. Acute encephalopathy associated with influenza and other viral infections. Acta Neurol Scand. 2007;115(4 Suppl):45–56.
Moro PL, Tepper NK, Grohskopf LA, et al. Safety of seasonal influenza and influenza A (H1N1) 2009 monovalent vaccines in pregnancy. Expert Rev Vaccines. 2012;11(8):911–21.
Mortimer PP. Was encephalitis lethargica a post-influenzal or some other phenomenon? Time to re-examine the problem. Epidemiol Infect. 2009;137(4):449–55.
Muhammad S, Haasbach E, Kotchourko M, Strigli A, Krenz A, Ridder DA, Vogel AB, Marti HH, Al-Abed Y, Planz O, et al. Influenza virus infection aggravates stroke outcome. Stroke. 2011;42(3):783–91.
Nakamura N, Nokura K, Zettsu T, Koga H, Tachi M, Terada M, Katoh H, Itoh Y, Osawa H, Ozeki T, et al. Neurologic complications associated with influenza vaccination: two adult cases. Intern Med. 2003;42(2):191–4.
Neurologic complications associated with novel influenza A (H1N1) virus infection in children - Dallas, Texas, May 2009. MMWR Morb Mortal Wkly Rep 2009, 58(28):773-778.
Newland JG, Laurich VM, Rosenquist AW, Heydon K, Licht DJ, Keren R, Zaoutis TE, Watson B, Hodinka RL, Coffin SE. Neurologic complications in children hospitalized with influenza: characteristics, incidence, and risk factors. J Pediatr. 2007;150(3):306–10.
Ng WF, Chiu SC, Lam DS, Wong YC, Tam S, Kwong NS, Loo KT, Yuen KY. A 7-year-old boy dying of acute encephalopathy. Brain Pathol. 2010;20(1):261–4.
Nichol KL, Nordin J, Mullooly J, Lask R, Fillbrandt K, Iwane M. Influenza vaccination and reduction in hospitalizations for cardiac disease and stroke among the elderly. N Engl J Med. 2003;348(14):1322–32.
Nunoi H, Mercado MR, Mizukami T, Okajima K, Morishima T, Sakata H, Nakayama S, Mori S, Hayashi M, Mori H, et al. Apoptosis under hypercytokinemia is a possible pathogenesis in influenza-associated encephalopathy. Pediatr Int. 2005;47(2):175–9.
Okabe N, Yamashita K, Taniguchi K, Inouye S. Influenza surveillance system of Japan and acute encephalitis and encephalopathy in the influenza season. Pediatr Int. 2000;42(2):187–91.
Okumura A, Abe S, Kidokoro H, Mizuguchi M. Acute necrotizing encephalopathy: a comparison between influenza and non-influenza cases. Microbiol Immunol. 2009;53(5):277–80.
Okumura A, Mizuguchi M, Kidokoro H, Tanaka M, Abe S, Hosoya M, Aiba H, Maegaki Y, Yamamoto H, Tanabe T, et al. Outcome of acute necrotizing encephalopathy in relation to treatment with corticosteroids and gammaglobulin. Brain Dev. 2009;31(3):221–7.
Olgar S, Ertugrul T, Nisli K, Aydin K, Caliskan M. Influenza a-associated acute necrotizing encephalopathy. Neuropediatrics. 2006;37(3):166–8.
Pandemic influenza: a priority for the neurological community. Lancet Neurol 2009; 8(10):869.
Parikh M, Dolson G, Ramanathan V, Sangsiraprapha W. Novel H1N1-associated rhabdomyolysis leading to acute renal failure. Clin Microbiol Infect. 2010;16(4):330–2.
Partinen M, Saarenpaa-Heikkila O, Ilveskoski I, et al. Increased incidence and clinical picture of childhood narcolepsy following the 2009 H1N1 pandemic vaccination campaign in Finland. PLoS ONE. 2012;7(3):33723.
Perez C, Loza E, Tinture T. Giant cell arteritis after influenza vaccination. Arch Intern Med. 2000;160(17):2677.
Preliminary results: surveillance for Guillain-Barre syndrome after receipt of influenza A (H1N1) 2009 monovalent vaccine - United States, 2009-2010. MMWR Morb Mortal Wkly Rep 2010, 59(21):657-661.
Reid AH, McCall S, Henry JM, Taubenberger JK. Experimenting on the past: the enigma of von Economo’s encephalitis lethargica. J Neuropathol Exp Neurol. 2001;60(7):663–70.
Ryan MM, Procopis PG, Ouvrier RA. Influenza A encephalitis with movement disorder. Pediatr Neurol. 1999;21(3):669–73.
Safety of influenza A (H1N1) 2009 monovalent vaccines - United States, October 1-November 24, 2009. MMWR Morb Mortal Wkly Rep 2009, 58(48):1351-1356.
Sakurai T, Kimura A, Tanaka Y, Hozumi I, Ogura S, Inuzuka T. Case of adult influenza type A virus-associated encephalopathy successfully treated with primary multidisciplinary treatments. Rinsho Shinkeigaku. 2007;47(10):639–43.
Sanchez-Torrent L, Trivino-Rodriguez M, Suero-Toledano P, Claret-Teruel G, Munoz-Almagro C, Martinez-Sanchez L, Jordan-Garcia I, Garcia–Garcia JJ. Novel influenza A (H1N1) encephalitis in a 3-month-old infant. Infection. 2010;38(3):227–9.
Sato S, Kumada S, Koji T, Okaniwa M. Reversible frontal lobe syndrome associated with influenza virus infection in children. Pediatr Neurol. 2000;22(4):318–21.
Shieh WJ, Blau DM, Denison AM, Deleon-Carnes M, Adem P, Bhatnagar J, Sumner J, Liu L, Patel M, Batten B, et al. 2009 pandemic influenza A (H1N1): pathology and pathogenesis of 100 fatal cases in the United States. Am J Pathol. 2010;177(1):166–75.
Shiomi M. Pathogenesis of acute encephalitis and acute encephalopathy. Nippon Rinsho. 2011;69(3):399–408.
Smeeth L, Thomas SL, Hall AJ, Hubbard R, Farrington P, Vallance P. Risk of myocardial infarction and stroke after acute infection or vaccination. N Engl J Med. 2004;351(25):2611–8.
Studahl M. Influenza virus and CNS manifestations. J Clin Virol. 2003;28(3):225–32.
Takanashi J. Two newly proposed infectious encephalitis/encephalopathy syndromes. Brain Dev. 2009;31(7):521–8.
Takanashi J, Tada H, Kuroki H, Barkovich AJ. Delirious behavior in influenza is associated with a reversible splenial lesion. Brain Dev. 2009;31(6):423–6.
Takanashi J, Tsuji M, Amemiya K, Tada H, Barkovich AJ. Mild influenza encephalopathy with biphasic seizures and late reduced diffusion. J Neurol Sci. 2007;256(1–2):86–9.
Tokuhira N, Shime N, Inoue M, et al. Mechanically ventilated children with 2009 pandemic influenza A/H1N1: results from the National Pediatric Intensive Care Registry in Japan. Pediatr Crit Care Med. 2012;13(5):e294–8.
Tsuji M, Yoshida T, Miyakoshi C, Haruta T. Is a reversible splenial lesion a sign of encephalopathy? Pediatr Neurol. 2009;41(2):143–5.
van der Vries E, Stelma FF, Boucher CA. Emergence of a multidrug-resistant pandemic influenza A (H1N1) virus. N Engl J Med. 2010;363(14):1381–2.
Vellozzi C, Broder KR, Haber P, Guh A, Nguyen M, Cano M, Lewis P, McNeil MM, Bryant M, Singleton J, et al. Adverse events following influenza A (H1N1) 2009 monovalent vaccines reported to the Vaccine Adverse Event Reporting System, United States, October 1, 2009-January 31, 2010. Vaccine. 2010;28(45):7248–55.
Watanabe T, Okazaki E, Shibuya H. Influenza A virus-associated encephalopathy with haemophagocytic syndrome. Eur J Pediatr. 2003;162(11):799–800.
Yamanouchi H, Kawaguchi N, Mori M, Imataka G, Yamagata T, Hashimoto T, Momoi MY, Eguchi M, Mizuguchi M. Acute infantile encephalopathy predominantly affecting the frontal lobes. Pediatr Neurol. 2006;34(2):93–100.
Yamanouchi H, Mizuguchi M. Acute infantile encephalopathy predominantly affecting the frontal lobes (AIEF): a novel clinical category and its tentative diagnostic criteria. Epilepsy Res. 2006;70(Suppl 1):S263–8.
Yang J, Wang Y-G, Xu Y-L, Ren X-L, Mao Y, Li X-W. A (H1N1) Influenza Pneumonia with Acute Disseminated Encephalomyelitis: a Case Report. Biomed Environ Sci. 2010;23(4):323–6.
Yao D, Mizuguchi H, Yamaguchi M, Yamada H, Chida J, Shikata K, Kido H. Thermal instability of compound variants of carnitine palmitoyltransferase II and impaired mitochondrial fuel utilization in influenza-associated encephalopathy. Hum Mutat. 2008;29(5):718–27.
Yokota S, Imagawa T, Miyamae T, Ito S, Nakajima S, Nezu A, Mori M. Hypothetical pathophysiology of acute encephalopathy and encephalitis related to influenza virus infection and hypothermia therapy. Pediatr Int. 2000;42(2):197–203.
Zinman L, Thoma J, Kwong JC, Kopp A, Stukel TA, Juurlink DN. Safety of influenza vaccination in patients with myasthenia gravis: a population-based study. Muscle Nerve. 2009;40(6):947–51.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tsai, J.P., Baker, A.J. Influenza-Associated Neurological Complications. Neurocrit Care 18, 118–130 (2013). https://doi.org/10.1007/s12028-012-9796-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12028-012-9796-8