 James P. Higham
James P. Higham- School of Physiology, Pharmacology and Neuroscience, University of Bristol, Bristol, United Kingdom
The cellular underpinnings of memory deficits in Alzheimer’s disease (AD) are poorly understood. We utilized the tractable neural circuits sub-serving memory in Drosophila to investigate the role of impaired Ca2+ handling in memory deficits caused by expression of human 0N4R isoform of tau which is associated with AD. Expression of tau in mushroom body neuropils, or a subset of mushroom body output neurons, led to impaired memory. By using the Ca2+ reporter GCaMP6f, we observed changes in Ca2+ signaling when tau was expressed in these neurons, an effect that could be blocked by the L-type Ca2+ channel antagonist nimodipine or reversed by RNAi knock-down of the L-type channel gene. The L-type Ca2+ channel itself is required for memory formation, however, RNAi knock-down of the L-type Ca2+ channel in neurons overexpressing human tau resulted in flies whose memory is restored to levels equivalent to wild-type. Expression data suggest that Drosophila L-type Ca2+ channel mRNA levels are increased upon tau expression in neurons, thus contributing to the effects observed on memory and intracellular Ca2+ homeostasis. Together, our Ca2+ imaging and memory experiments suggest that expression of the 0N4R isoform of human tau increases the number of L-type Ca2+ channels in the membrane resulting in changes in neuronal excitability that can be ameliorated by RNAi knockdown or pharmacological blockade of L-type Ca2+ channels. This highlights a role for L-type Ca2+ channels in tauopathy and their potential as a therapeutic target for AD.
Introduction
The accumulation of the microtubule-associated protein tau (MAPT) within central neurons is a key histopathological feature of Alzheimer’s disease (AD) (Kaufman et al., 2018; Nisbet and Gotz, 2018). Post-mortem AD brain samples contain the hallmark accumulation of extracellular amyloid beta (Aβ) plaques and intracellular neurofibrillary tangles (NFTs) of hyperphosphorylated tau, resulting in neurodegeneration and brain atrophy. The accumulation of tau is more correlated with the progression of AD pathology and symptoms than the magnitude of Aβ plaques (Arendt et al., 2016). However, the mechanisms by which tau disrupts neuronal function, and consequently behavior, are not clear. Tau exists in six major isoforms formed by alternative splicing of exons 2 and/or 3 (0N, 1N or 2N) and 10 (3R or 4R) of the MAPT-17 gene (Buee et al., 2000). There is some evidence that 4R isoforms are upregulated in post-mortem brains, particularly within the hippocampus of AD patients (Boutajangout et al., 2004; Espinoza et al., 2008; Hasegawa et al., 2014), and show stronger affinity binding to tubulin and aggregation than 3R forms (Arendt et al., 2016). However, both 3R and 4R isoforms are present in NFTs, suggesting both play a role in pathology. The expression of 0N4R tau in model organisms can yield neuronal and behavioral dysfunction prior to the onset of neurodegeneration (Wittmann et al., 2001; Mershin et al., 2004). This suggests that tau’s detrimental effect on neurons is sufficient to drive behavioral changes before neuronal death.
Patients with AD display memory impairment, and once the onset of neurodegeneration has occurred, treating the symptoms of AD becomes exceedingly difficult, so targeting the earlier signs of neuronal dysfunction is likely to be more efficacious. To this end, an understanding of how tau influences neuronal function is required. Disrupted calcium (Ca2+) homeostasis including increased Ca2+ levels causing excitotoxicity has been posited as a key pathophysiology in AD, which may underpin the early stages of disease and precipitate neurodegeneration (Mattson and Chan, 2001; Brzyska and Elbaum, 2003; Canzoniero and Snider, 2005). Recent work suggests that these changes in neuronal excitability and Ca2+ signaling provide an important link between Aβ and tau pathology and disease progression (Spires-Jones and Hyman, 2014; Wu et al., 2016), but exactly how remains unknown. Different rodent models of tauopathy recapitulate some features of AD pathology including changes in excitability (Booth et al., 2016a, b), increased Ca2+ signaling (Wang and Mattson, 2014), neurodegeneration (Spillantini and Goedert, 2013), and impaired synaptic plasticity and memory (Arendt et al., 2016; Biundo et al., 2018). It is not clear what the source of increased Ca2+ is, however, increased levels of Ca2+ channels, such as the L-type voltage-gated Ca2+ channel (CaV1), have been shown to be upregulated in rodent models of AD, with blockers, such as nifedipine, being effective in trails to prevent the cognitive decline that occurs in AD (Coon et al., 1999; Anekonda et al., 2011; Goodison et al., 2012; Nimmrich and Eckert, 2013; Daschil et al., 2015). The role of voltage-gated Ca2+ channels in memory or AD has not been studied in Drosophila. Drosophila contains three different voltage-gated Ca2+ channel genes including the DmCa1D or Ca-α1D gene that encodes a high voltage-activated current and is equivalent to the vertebrate CaV1.1-1.4 genes that encode L-type Ca2+ channels (Worrell and Levine, 2008).
Alzheimer’s disease and tauopathies have been modeled in Drosophila (Iijima-Ando and Iijima, 2010; Wentzell and Kretzschmar, 2010; Papanikolopoulou and Skoulakis, 2011), with targeted neuronal expression of human 0N4R tau causing neurodegeneration, shortened lifespan, circadian, sleep and motor deficits (Wittmann et al., 2001; Kerr et al., 2011; Sealey et al., 2017; Buhl et al., 2019; Higham et al., 2019). Furthermore, during development at the larval neuromuscular junction, motor neurons overexpressing human 0N3R or 0N4R caused a reduction in size and irregular and abnormally shaped synaptic terminals, a reduction in endocytosis and exocytosis and a reduction in high frequency synaptic transmission (Chee et al., 2005; Zhou et al., 2017). Also, while expression of tau 0N3R in the adult giant fiber system caused increased failure rate at high frequency stimulation, expression of 0N4R caused defects in stimulus conduction, response speed and conduction velocity (Kadas et al., 2019).
Learning and memory deficits can also be assessed in Drosophila using aversive olfactory classical conditioning (Malik et al., 2013; Malik and Hodge, 2014). Different mushroom body (MB) neuropils underpin specific phases of memory and are redundant during others (Pascual and Preat, 2001; Krashes et al., 2007; Davis, 2011). MB neurons send axons that terminate in lobed structures, with memory acquisition being mediated by the α′β′ lobe neurons and memory-storage by the αβγ neurons. In addition, a number of additional pairs of neurons innervate or are innervated by the MB and mediate different aspects of memory. For instance, the amnesiac expressing dorsal paired medial (DPM) and anterior paired lateral (APL) neurons are thought to consolidate and stabilize labile memory (Pitman et al., 2011). Finally, the MB innervate a pair of M4/6 MB output neurons via a cholinergic synapse, with optogenetic manipulation of the M4/6 neurons switching between appetitive and aversive memory (Barnstedt et al., 2016). Previous work has demonstrated that expression of tau in γ neurons caused a reduction in learning and 1.5 h memory in 3–5 days old young flies which were shown to still have their γ neurons intact, prior to their degeneration around day 45 (Mershin et al., 2004). Moreover, pan-neuronal 0N4R expression caused learning and long-term memory loss while 0N3R tau expression failed to do so (Sealey et al., 2017).
Therefore, Drosophila expressing human tau can be used to study AD relevant behaviors and their underlying neuronal mechanism, with loss of memory being possible prior to neurodegeneration. Although tau-mediated changes in neuronal properties are likely underpinning the memory impairment observed in Drosophila AD models, these changes have not been extensively studied in Drosophila MB neurons.
In this study, we determined the effect of human 0N4R tau expression in different neuropils of the Drosophila memory circuit on one-hour memory. We measured the effect of 0N4R tau on MB output neuron mediated memory and Ca2+ signaling and described a potential interaction between 0N4R tau with L-type voltage-gated Ca2+ channels which may underpin memory impairment.
Results
To determine the effect of human tau 0N4R on memory, we targeted expression of the transgene to different neuronal populations of the Drosophila memory circuit and performed one-hour olfactory aversive conditioning. Driving expression of a human tau 0N4R transgene (Wittmann et al., 2001; Kerr et al., 2011; Higham et al., 2019) in the entire MB (OK107-Gal4 (Malik et al., 2013), pGal4 = 0.004, pUAS = 0.01, Figure 1A) yielded memory deficient flies. Similarly, when expressed in the memory-relevant M4/6 MB output neurons (Barnstedt et al., 2016), R21D02-Gal4, GCaMP6f > tau flies displayed greatly reduced memory performance compared to control counterparts (pGal4 = 0.02, pUAS < 0.001, Figure 1B). We also found that tau caused a significant reduction in one-hour memory when expressed in the α′β′ MB neurons (c305a-Gal4 (Krashes et al., 2007), pGal4 = 0.02, pUAS = 0.01, Figure 1C), which mediate memory acquisition and in the memory-storing αβγ neurons (MB247-Gal4 (Krashes et al., 2007), pGal4 < 0.001, pUAS = 0.003) and dorsal paired medial (DPM) neurons (amn(c316)-Gal4, pGal4 = 0.002, pUAS = 0.03), which consolidate and stabilize labile memory (Pitman et al., 2011). All animals used for aversive conditioning were aged 2–5 days and displayed naïve avoidance of shock (Supplementary Table S1). Likewise, all genotypes showed similar naïve avoidance of the odors used for olfactory conditioning (Supplementary Table S1). Thus, all genotypes were able to detect both odors and shock, verifying that memory defects were bone fide and not attributable to a sensorimotor defect.
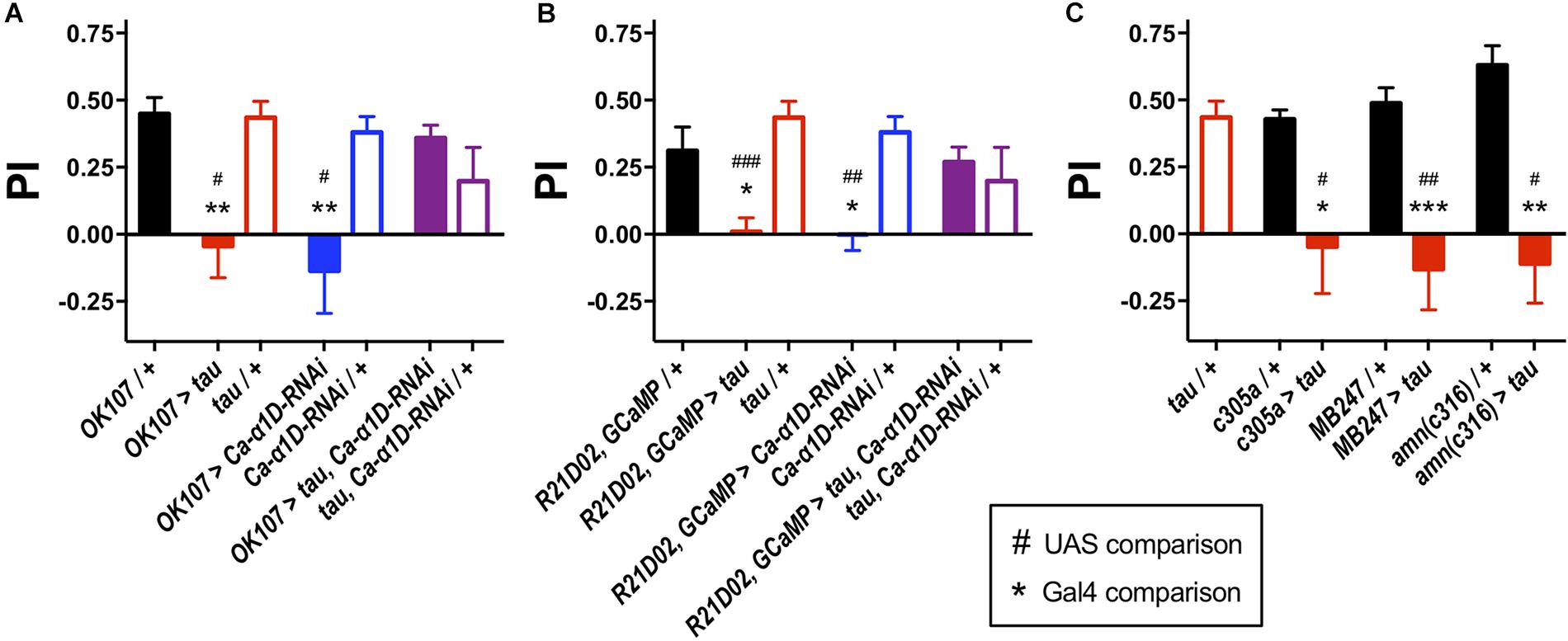
Figure 1. Knock-down of L-type Ca2+ channels rescued tau-induced memory defects. (A) Tau expression in the MB (OK107-Gal4) ablated one-hour memory (n = 7 experiments with 50–100 flies each, red bar), as did the expression of an RNAi to the gene encoding the L-type Ca2+ channel alpha subunit (Ca-α1D-RNAi, n = 8, blue bar) compared to both Gal4 (n = 12, black bar) and UAS controls (tau, n = 11, open red bars; Ca-α1D-RNAi, n = 9, open blue bars). Co-expression of both transgenes (n = 12, purple bar) resulted in flies with a memory performance equivalent to that of control animals, as did the UAS controls (n = 4, open purple bar). (B) Likewise, expression of tau (n = 9) or Ca-α1D-RNAi (n = 6) in the MB efferent M4/6 neurons (R21D02-Gal4, n = 10) abolished memory. Memory was restored to wild-type levels by co-expression of both transgenes (n = 15). (C) Expression of tau in neuronal subpopulations of the MB impaired memory. Expression was driven in the α′β′ neurons (c305a-Gal4, n = 6 control, n = 4 tau), αβγ neurons (MB247-Gal4, n = 10 control, n = 7 tau) and dorsal paired medial neurons (amn(c316)-Gal4, n = 5 control, n = 4 tau). Data analyzed with Kruskal–Wallis test with Dunn’s post hoc tests (A,C) or with one-way ANOVA with Dunnett’s post hoc tests (B). */#p < 0.05, **/##p < 0.01, and ***/###p < 0.001. Note that UAS/ + controls are the same for each panel.
To assess changes in Ca2+ handling which may underpin behavioral dysfunction, we imaged the M4/6 neurons in whole ex vivo brains using GCaMP6f as these neurons are pertinent to memory and single neurons can be visualized (Barnstedt et al., 2016). There did not appear to be a difference in baseline fluorescence of these neurons between control and tau-expressing animals (p = 0.5, Figure 2A), suggesting no effect on basal Ca2+ handling, assuming comparable GCaMP6f expression between genotypes. Bath application of high potassium chloride (KCl) concentration resulted in a robust and transient elevation in fluorescence which could be ameliorated by removing Ca2+ from the external solution (12.8 ± 1.0% of control, p = 0.003) or by adding cadmium (200 μM, 11.75 ± 11.75% of control, p = 0.009), a general blocker of Drosophila voltage-gated Ca2+ channels (Ryglewski et al., 2012), to the external solution (Supplementary Figure S2A). This indicated that the transient relies upon Ca2+ influx through voltage-gated Ca2+ channels, or that cadmium blocked presynaptic Ca2+ channels and, consequently, neurotransmitter release and activation of M4/6 neurons.
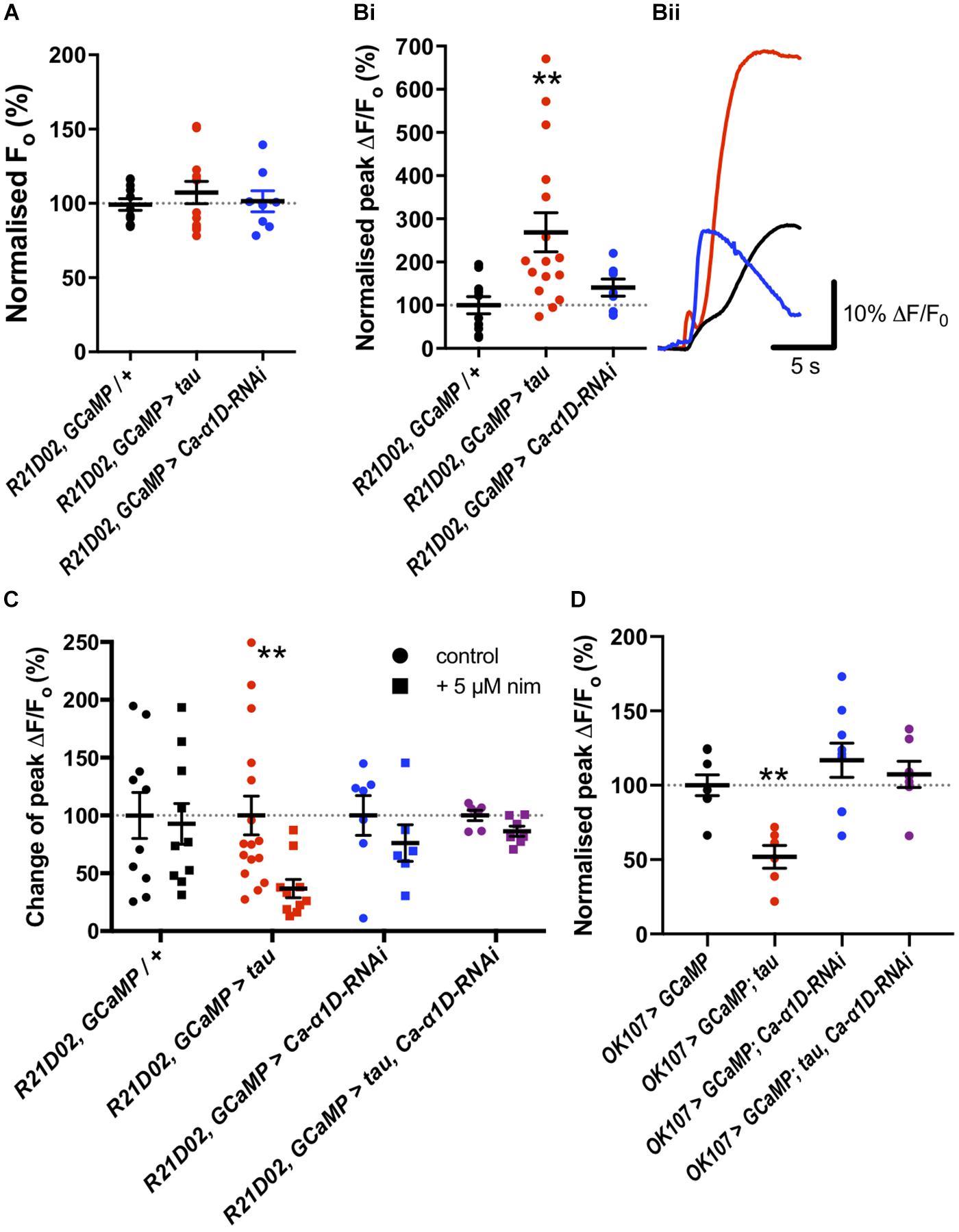
Figure 2. Tau expression elevated nimodipine-sensitive Ca2+ channels in mushroom body (MB) and M4/6 neurons. (A) Basal Ca2+ levels were no different between control neurons and those expressing tau or Ca-α1D-RNAi. (Bi) However, the peak magnitude of Ca2+ transients were greater in the tau-expressing neurons than in control or Ca-α1D-RNAi-expressing neurons. (Bii) GCaMP6f relative fluorescence changes over time traces of M4/6 neurons in response to 100 mM KCl for control (black trace), tau (red trace) and Ca-α1D-RNAi (blue trace). (C) Ca2+ transients in control and Ca-α1D-RNAi-expressing neurons were not sensitive to the L-type Ca2+ channel blocker nimodipine (black and blue symbols, respectively). Driving tau expression in M4/6 neurons conferred sensitivity of Ca2+ transients to nimodipine (red symbols). The effect of tau on Ca2+ transient nimodipine sensitivity was ablated by co-expression of tau and Ca-α1D-RNAi in these neurons (purple symbols). Control for each genotype normalized to 100%. (D) Tau expression in the MB reduced peak fluorescence following a high KCl challenge; an effect reversed by co-expression of Ca-α1D-RNAi. Data analyzed with one-way ANOVA with Dunnett’s post hoc tests panels (A,D), with Kruskal–Wallis test with Dunn’s post hoc tests panel (B) or with two-way ANOVA with Sidak’s post hoc tests panel (C). */#p < 0.05, **/##p < 0.01, and ***/###p < 0.001. Note that data for OK107 > GCaMP and OK107 > GCaMP, tau are reproduced from Higham et al. (2019).
KCl-evoked Ca2+ transients in tau-expressing M4/6 neurons were almost three-fold greater than in control neurons (268.9 ± 45.2% of control, p = 0.005, Figure 2B). Since previous reports have documented the potential involvement of L-type Ca2+ channels in AD (Wang and Mattson, 2014; Coon et al., 1999), we tested whether the change in magnitude of Ca2+ transients in M4/6 neurons was dependent on these channels by applying the clinically-used L-type channel-selective blocker nimodipine (Nimmrich and Eckert, 2013; Terada et al., 2016). The addition of 5 μM nimodipine to the external solution had no effect on the magnitude of Ca2+ transients in control brains (p > 0.9, Figure 2C). However, the peak of the transient in tau-expressing neurons was sensitive to nimodipine and was reduced to a magnitude indistinguishable from control (98.9 ± 21.1% of control, p > 0.9). The elevated Ca2+ influx seen in these neurons may, therefore, be due to augmented L-type Ca2+ channels. To further investigate which type of voltage-gated Ca2+ channel mediated the Ca2+ influx, we also tested the effect of addition of the L-type and T-type Ca2+ channel blocker, amiloride (1 mM), to nimodipine on the KCl-induced Ca2+ transient, as this compound greatly reduced Ca2+ currents in cultured embryonic Drosophila giant neurons (Peng and Wu, 2007). Amiloride did not significantly reduce the peak of the Ca2+ transient in M4/6 neurons (p = 0.2, Supplementary Figure S2B). The lack of effect of amiloride likely indicates developmental or cell-specific differences in Ca2+ channel expression or that the voltage-gated Ca2+ channels blocked by amiloride largely overlap with nimodipine which do not contribute significantly to the Ca2+ transient in wild-type M4/6 neurons.
To corroborate our pharmacological data, we tested the nimodipine sensitivity of Ca2+ transients in M4/6 neurons co-expressing tau and an RNAi to the L-type Ca2+ channel gene (UAS-Ca-α1D-RNAi), that has been shown to reduce L-type Ca2+ channel currents and protein levels by over 75% in Drosophila neurons (Kadas et al., 2017). Ca2+ transients in these neurons displayed a much reduced, and statistically insignificant (p > 0.05, Figure 2C), block by nimodipine compared to transients in neurons expressing tau alone (13.2% vs. 62.3% reduction in peak). This data shows that the elevated Ca2+ transients in tau-expressing M4/6 neurons rely on L-type Ca2+ channels. As L-type channels negatively regulate neuronal excitability (Worrell and Levine, 2008), RNAi mediated reduction of L-type Ca2+ channels increases neuronal activity (Kadas et al., 2017).
We went on to test the involvement of the L-type Ca2+ channel itself in memory by knocking down its expression with RNAi. Knock-down of Ca-α1D in the M4/6 neurons abolished one-hour memory (pGal4 = 0.03, pUAS = 0.007, Figure 1B). These animals exhibited no sensorimotor defects (Supplementary Table S1), verifying the observed phenotype as a genuine memory defect. In alignment with the lack of effect of nimodipine on the Ca2+ transient in control neurons, knock-down of the L-type channel had no effect on KCl-evoked Ca2+ influx in M4/6 cells (p = 0.6, Figure 2B).
We sought to resolve whether there was a behavioral interaction between 0N4R tau and the L-type Ca2+ channel by testing whether knocking down Ca-α1D could ameliorate the effect of tau on memory as it did on Ca2+ signaling. Strikingly, even though expression of human tau or Ca-α1D-RNAi removed memory alone, co-expression in M4/6 neurons yielded flies with memory performance indistinguishable from wild-type animals (pGal4 > 0.9, pUAS > 0.9, Figure 1B). We tested whether this restoration of memory could have been a consequence of Gal4 dilution due to the presence of multiple transgenes. Expression of an innocuous gene, UAS-GFP, with either UAS-tau or UAS-Ca-α1D-RNAi yielded animals with impaired memory (p = 0.02 and 0.005, respectively, Supplementary Figure S1). This demonstrates that dilution of transgene expression is not responsible for the observed change in memory performance. Lastly, the expression of transgenes in the M4/6 output neurons had no effect on the animals’ naïve sensorimotor behavior indicating that the effects seen in memory are not due to defective sensory responses (Supplementary Table S1).
To test if the apparent interaction between tau and L-type Ca2+ channels occurred in other neurons, we performed Ca2+ imaging of the entire MB (OK107-Gal4). The KCl-evoked Ca2+ response in this large population of neurons was reduced by the expression of tau (48.1% reduction, p = 0.006, Figure 2D; Higham et al., 2019), likely reflecting a reduction in excitability. MB neuronal architecture appeared grossly intact in tau-expressing animals (data not shown), and neurodegeneration has been shown not to play a significant role in memory defects at this age (Higham et al., 2019). In alignment with observations in M4/6 neurons, the expression of Ca-α1D-RNAi in the MB abolished one-hour memory (pGal4 = 0.001, pUAS = 0.04, Figure 1A) but did not affect the magnitude of the Ca2+ transient in these neurons (p = 0.4, Figure 2D). The co-expression of these two transgenes in the MB resulted in a Ca2+ transient which was no different in magnitude compared to control animals (p > 0.9, Figure 2D). The ablation of one-hour memory caused by tau or Ca-α1D-RNAi expression in MB neurons was also reversed by co-expression of both transgenes as these animals exhibited a memory performance equivalent to control animals (pGal4 > 0.9, pUAS > 0.9, Figure 1A).
The mechanism underlying tau-mediated augmentation of Ca2+ influx through L-type channels is not clear. It could be due to elevated expression of the Ca-α1D gene, increased trafficking to, or reduced recycling from, the plasma membrane or changes in single channel properties such as conductance or open probability. To investigate this further, we measured Ca-α1D expression in whole brain extracts by RT-qPCR. All transgenes were expressed in all neurons (elav-Gal4) to ensure changes in gene expression were detectable (Figure 3). Ca-α1D-RNAi knock-down reduced expression of the channel by 58% (p < 0.05), while expression of tau lead to a 68% (p = 0.04) increase in Ca-α1D. Interestingly, co-expression of tau and Ca-α1D-RNAi restored Ca-α1D to 72% (p = 0.3) of control fly levels. This strongly suggests that the effects observed in behavior and Ca2+ imaging are due to a change in Ca-α1D expression in the brain.
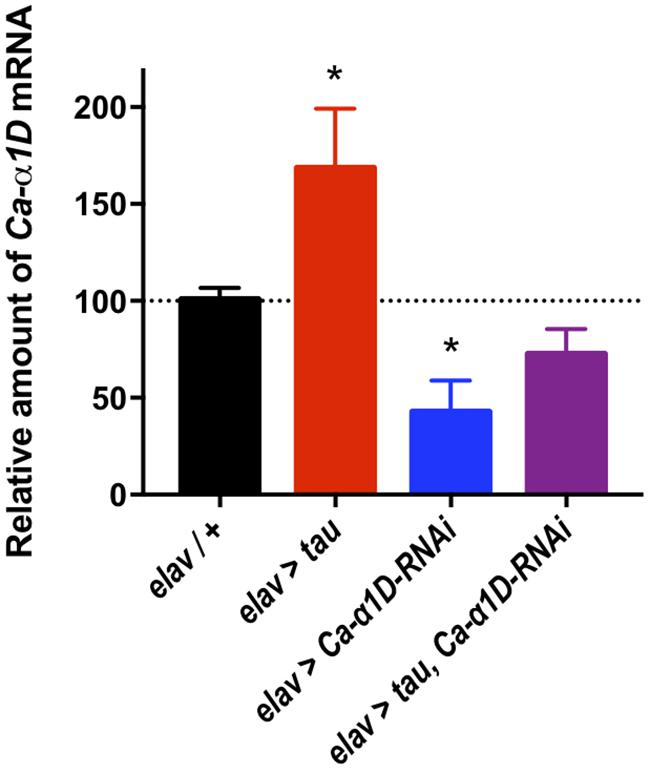
Figure 3. Tau expression increased Ca2+ channelα1D mRNA levels. Pan-neuronal Tau expression led to a 68% increase in Ca-α1D mRNA levels compared to controls, while expressing a RNAi against Ca-α1D reduced its expression by 58%. Co-expression of the RNAi in tau expressing flies partially restored levels of Ca-α1D compared to controls. n = 6 biological replicates of 23 or more heads each for all groups. Data analyzed with one-way ANOVA with Holm-Sidak’s post hoc tests. ∗p < 0.05.
Discussion
Selectively expressing tau in MB neuronal subsets revealed a non-specific adverse effect on memory processing (Figure 1). MB neurons sub-serving specific memory phases rely on different signaling molecules. For example, CaMKII is required in α′β′ neurons, but not αβγ, and vice versa for KCNQ channels (Cavaliere et al., 2013; Malik et al., 2013). This suggests that tau either promiscuously interacts with and disrupts numerous intracellular components or disrupts a pathway common to all MB neurons. In vivo Ca2+ imaging of different MB neuronal subsets has revealed the importance of Ca2+ transient plasticity in olfactory associative memory (Yu et al., 2006; Sejourne et al., 2011). Following conditioning, MB neurons exhibit differential Ca2+ responses to the CS+ and CS- odors (Yu et al., 2006; Sejourne et al., 2011). In the β′ lobe dendrites of M4/6 neurons, exposure to an aversively conditioned CS+ odour results in a greater Ca2+ influx compared with CS-, with the reverse being true for appetitively conditioned odors (Owald et al., 2015). Other MB neurons display a similar distinction between CS+ and CS-, with these differences believed to coordinate avoidance or approach behavior. Given that tau expression aberrantly elevated stimulus-evoked Ca2+ influx, it is plausible that this interferes with conditioning-induced Ca2+ transient plasticity. Augmented Ca2+ entry via L-type channels in tau-expressing neurons may ablate the difference in Ca2+ influx between CS+ and CS-, rendering them indistinguishable at the cellular level.
The knock-down of the Drosophila L-type Ca2+ channel demonstrated its importance in both the MB and the M4/6 neurons for memory. This aligns with data from Cav1.2 knock-out mice, which are deficient in spatial memory tasks (Moosmang et al., 2005; White et al., 2008). Despite R21D02, GCaMP6f > Ca-α1D-RNAi and OK107 > Ca-α1D-RNAi flies being memory deficient, the M4/6 and MB neurons of these animals displayed no reduction in evoked Ca2+ entry, nor did nimodipine have any effect on the Ca2+ transient in control M4/6 neurons (Figure 2). This suggests that perhaps only a small population of L-type channels is present in these neurons. A previous electrophysiological study of Ca2+ currents in cultured Drosophila giant embryonic neurons revealed only a very small block by nifedipine, likely reflecting a low number of L-type channels (Peng and Wu, 2007). Likewise an electrophysiological and pharmacological study of the adult OK107-Gal4 MB neurons again showed only a small contribution of L-type channels to their Ca2+ transients (Jiang et al., 2005). Fluorescence imaging using GCaMP may not be sufficiently sensitive to resolve such a small contribution to global Ca2+ influx – a contribution that is nonetheless important for cellular function and, hence, memory.
It is not known whether the L-type channel plays any role in the plasticity of Ca2+ transients in Drosophila per se. However, these channels are vital for the function of memory-associated neurons. The generation of medium and slow afterhyperpolarizations (AHPs), which are a period of prolonged hyperpolarized membrane potential following action potential firing, in hippocampal neurons is dependent on Ca2+ entry through L-type channels (Lancaster and Adams, 1986; Marrion and Tavalin, 1998; Bowden et al., 2001). Elevated Ca2+ entry would augment AHPs and suppress neuronal firing, thereby disrupting neural circuit function and the behavior subserved by that circuit. This is apparent in mice and rabbits, which exhibit an age-related memory decline with concomitant elevation of L-type Ca2+ channel expression and AHP magnitude in the hippocampus (Moyer et al., 1992; Nunez-Santana et al., 2014). This impairs stimulus-evoked changes in neuronal activity which underpin learning (Moyer et al., 2000). Blocking augmented AHPs in aged rabbits with nimodipine facilitated enhanced performance in associative learning tasks (Straube et al., 1990). L-type Ca2+ channels also negatively regulate neuronal excitability in Drosophila neurons (Worrell and Levine, 2008), and so their augmentation by tau may exert a similar effect on Drosophila neurons to that observed in aging mammals, with their knock-down resulting in increased firing of neurons (Kadas et al., 2017).
In M4/6 neurons, we observed raised evoked Ca2+ influx due to tau expression, while Ca2+ influx was suppressed in MB neurons (Figures 2B,D). Augmentation of the Ca2+ transient in single tau-expressing M4/6 neurons reflects the elevated Ca2+ influx through L-type channels, as the transients were reduced by nimodipine and L-type channel knock-down. However, in a large population of MB neurons (∼2500 neurons) the elevated Ca2+ influx through L-type channels is likely small relative to the reduced Ca2+ levels due to suppressed neuronal activity, therefore a reduction in the Ca2+ transient was observed. The reduced excitability of MB neurons was rescued by co-expression of Ca-α1D-RNAi as this manipulation opposes the suppression of excitability caused by tau (Kadas et al., 2017). Not only were the tau-induced Ca2+ signaling defects rescued by manipulation of L-type Ca2+ channels, but so was olfactory memory (Figures 1A,B). This lends further credence to the suggestion that tau interacts with the L-type Ca2+ channel and also shows that this interaction solely mediates memory dysfunction, at least in young animals.
Raised L-type Ca2+ channel expression has also been documented in other AD models, as well as in aging. In agreement with our data, L-type Ca2+ current density was elevated in CA1 neurons of 3 × Tg mice (Wang and Mattson, 2014), as were AHPs in the dorsomedial entorhinal cortex of rTg4510 mice (Booth et al., 2016a). Our data shows that wild-type 0N4R tau, as well as the frontotemporal dementia-associated P301L mutant expressed in 3 × Tg and rTg4510 mice, is capable of augmenting Ca2+ influx in neurons. What is more, elevated expression of a mammalian L-type Ca2+ channel (CaV1.2) was observed in a neuroblastoma cell line following transgenic Aβ42 expression; this resulted in reduced cell viability and six-fold increase in Ca2+ influx, that could be ameliorated by the L-type Ca2+ channel dihydropyridine blockers, nimodipine and isradipine (Copenhaver et al., 2011). This study went on to show that the L-type channel blocker, isradipine could increase survival of Drosophila overexpressing human amyloid precursor protein (APP695), as well as decreasing the accumulation of Aβ and phosphorylated tau in the triple transgenic AD mice (3 × TgAD) which express human Presenilin 1M146V, APPSwedish and tauP30L. This indicates that correcting defective Ca2+ handling in AD may be of therapeutic benefit, particularly as L-type Ca2+ channels appear to be relevant in the human AD brain, too.
Raised L-type Ca2+ channel expression has been documented in the brains of AD patients (Coon et al., 1999), with increased L-type channels thought to underlie the memory loss and neurodegeneration that occurs in dementia (Missiaen et al., 2000; Mattson and Chan, 2001; Canzoniero and Snider, 2005; Thibault et al., 2007). Our data shows that there is increased expression of the Drosophila L-type Ca2+ channel, Ca-α1D, in tau expressing neurons, therefore suggesting conserved mechanisms of Aβ and tau-related calcium deficits across species (Figure 3). Importantly, clinical trials of L-type blockers show a slowing of cognitive decline in AD patients (Anekonda et al., 2011; Goodison et al., 2012; Nimmrich and Eckert, 2013).
In summary, using behavioral, physiological, pharmacological and molecular methods, we show that knock-down of the Drosophila L-type Ca2+ channel Ca-α1D can rescue tau mediated olfactory learning deficits by restoring Ca2+ handling in MB neurons.
Materials and Methods
Drosophila Genetics
Flies were raised at a standard density with a 12 h:12 h light dark (LD) cycle on standard Drosophila medium (0.7% agar, 1.0% soya flour, 8.0% polenta/maize, 1.8% yeast, 8.0% malt extract, 4.0% molasses, 0.8% propionic acid, and 2.3% nipagen) at 25°C. Wild-type control was Canton S w- (CSw-) and R21D02-GAL4, UAS-GCaMP6f were kind gifts from Prof. Scott Waddell (University of Oxford). UAS-human MAPT (TAU 0N4R) wild-type (Wittmann et al., 2001; Kerr et al., 2011) was a kind gift from Prof. Linda Partridge (University College London), UAS-GFP was a gift from Prof. Mark Wu (John Hopkins University). The following flies were obtained from the Bloomington and Vienna fly stock centers: OK107-Gal4 (Bloomington Drosophila stock center number BDSC:854), c305a-Gal4 (BDSC:30829), amn(c316)-Gal4 (BDSC:30830), MB247-Gal4 (BDSC:50742), elav-Gal4 (BDSC:8760), UAS-GCaMP6f (BDSC:42747) and UAS-Ca-α1D-RNAi flies [Vienna Drosophila resource center GD51491 (Kadas et al., 2017)].
Aversive Olfactory Conditioning
All memory experiments were carried out at 25°C and 70% relative humidity under dim red light. Flies were used for experiments after 2–5 days of aging at 25°C and 70% relative humidity in a 12-hour light: 12-hour dark environment. Using a previously published protocol (Mershin et al., 2004; Krashes et al., 2007; Kosmidis et al., 2010; Malik et al., 2013; Malik and Hodge, 2014; Barnstedt et al., 2016), groups of 25–50 flies were first transferred from food tubes into the training tube lined with an electrifiable grid. After acclimatization to the electrified tube for 90 s, flies were exposed to either 3-octanol (OCT, Sigma) or 4-methylcyclohexanol (MCH, Sigma) (conditioned stimulus, CS+) paired with twelve 70 V DC electric shocks (unconditioned stimulus, United States) over 60 s (shocks of duration 1.25 s with inter-shock latency of 3.75 s). This was followed by a 30 s rest period with no stimulus. Flies were then exposed to the reciprocal odour (CS−) for 60 s with no electric shock. Memory retention was tested one-hour post-conditioning (intermediate-term memory). To account for any innate bias the flies may have towards an odour, the CS + odour was reversed in alternate groups of flies and the performances from these two groups averaged to give n = 1. Moreover, the order of delivery of CS+ and CS− was alternately reversed.
To test memory, flies were placed at the choice point of a T-maze with one pathway exposed to CS+ and the other to CS−. After 120 s, the number of flies choosing each pathway was counted. Memory was quantified using the performance index (PI):
where NCS– and NCS+ is the number of flies choosing CS− and CS+, respectively. A PI = 1 indicates perfect learning where all flies chose CS−, and PI = 0 indicates a 50:50 split between CS− and CS+ and, therefore, no learning.
Calcium Imaging
GCaMP imaging was performed using previously published protocols (Cavaliere et al., 2012; Gillespie and Hodge, 2013; Malik et al., 2013; Schlichting et al., 2016; Shaw et al., 2018) with flies being anaesthetized on CO2, decapitated and their brains dissected out of the head in extracellular saline containing (in mM): 101 NaCl, 1 CaCl2, 4 MgCl2, 3 KCl, 5 D-glucose, 1.25 NaH2PO4, and 20.7 NaHCO3 adjusted to a final pH of pH 7.2 with an osmolality of 247–253 mmol/kg. Brains were held ventral side up in a recording chamber using a custom-made anchor and visualized with a 40× water-immersion lens on an upright microscope (Zeiss Examiner Z1).
Brains were superfused with extracellular saline (5 mL/min) as above and cells were depolarized by bath application of 100 mM KCl in extracellular solution (362 mmol/kg) for 15 s at 5 mL/min. Drug-containing or Ca2+-lacking solutions were superfused over the brain for 60 s prior to imaging. The Ca2+-lacking external solution contained 8 mM MgCl2.
Images were acquired at 8 Hz with 50 ms exposure using a CCD camera (ZEISS Axiocam) and a 470 nm LED light source (Colibri, ZEISS) and recorded with ZEN (Zeiss, 4 frames/sec). Baseline fluorescence (F0) was taken as the mean fluorescence during the 10 s (80 images) prior to the start of KCl perfusion. The change in fluorescence relative to baseline [(F-F0)/F0, where F is fluorescence at a given time] following KCl addition was recorded, and the peak change [(Fmax-F0)/F0] was used as a metric of transient [Ca2+] increase. Example traces were plotted using Origin 9 (Origin Lab).
All chemicals were purchased from Sigma-Aldrich (Gillingham, United Kingdom), except for nimodipine which was purchased from Tocris (Bristol, United Kingdom) and amiloride which was donated by Prof. David Sheppard (University of Bristol).
RT-qPCR
Relative measure of Ca-α1D expression levels was assessed by two-step qPCR. 2–5 days old male flies were anesthetized with CO2 and decapitated, obtaining six biological replicates with 23 heads each. Total RNA was extracted from head lysates by organic phenol/chloroform method using TRIzol reagent (Invitrogen). RNA quantification was carried out in Nanodrop spectrophotometer (Thermo Scientific) and RNA integrity was checked by electrophoresis in 1% agarose gel. Samples were treated with TURBO DNA-free kit (Invitrogen) in order to remove genomic DNA contamination. Reverse transcription was carried out using RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific) following manufacturer’s instructions, with 500 ng of RNA as template and Oligo (dT) as primer to amplify total mRNA. cDNA samples were stored at −20°C or used immediately for qPCR reactions.
Quantitative PCR reactions were carried out in QuantStudio 3 Real-Time PCR system (Applied Biosystems) using HOT FIREPol EvaGreen qPCR Mix Plus (Solis BioDyne). The primers used to amplify Ca-α1D mRNA were as follows: Ca1DF 5′-CCTTGAGGGCTGGACTGATG-3′ and Ca1DR 5′-ATCACGAAGAAGGCACCCAG-3′ with a PCR product expected size of 108 bp and 104% primer efficiency (Supplementary Figure S3). As a housekeeping gene, the following primers for GAPDH2 mRNA were used: GAPDH2F 5′-CGTTCATGCCACCACCGCTA-3′ and GAPDH2R 5′-CCACGTCCATCACGCCACAA-3′. The expected PCR product size was 72 bp and the primer efficiency was 100%. To activate DNA polymerase, a first step of 15 min at 95°C was used, followed by 50 cycles of 30 s at 95°C, 30 s at 60°C, followed by a 1 min 72°C elongation step. At the end of the experiment, a temperature ramp from 60°C to 95°C was used for melting curve analysis and the product fit to the predicted melting curve obtained by uMelt software (Dwight et al., 2011). Quantification for each genotype and each gene was carried out using the 2(–Δ Δ Ct) method and data was expressed as a percentage of change.
Analysis
All data were analyzed using Prism 7 (GraphPad Inc.). Data were scrutinized to check they met the assumptions of parametric statistical tests, and non-parametric, rank-based alternatives were used where appropriate. Details of statistical tests used are in figure legends. Data are presented as mean ± standard error of mean.
Data Availability
The raw data supporting the conclusion of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
All authors devised and performed the experiments, and wrote and edited the manuscript. JH secured the funding.
Funding
This work was supported by a CONICYT-PCHA/Doctorado Nacional/2016-21161611 studentship to SH. GW4 accelerator (GW4-AF2-002), Alzheimer’s Research United Kingdom network grant, Alzheimer’s Society undergraduate grants and Leverhulme Trust grant (RPG-2016-318) to JH. The funders had no other part in research.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Drs. Linda Partridge, Scott Waddell, and Mark Wu for providing the fly stocks, and Drs. Kofan Chen, Bilal Malik, and Neil Marrion for helpful advice and comments on the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2019.00409/full#supplementary-material
References
Anekonda, T. S., Quinn, J. F., Harris, C., Frahler, K., Wadsworth, T. L., and Woltjer, R. L. (2011). L-type voltage-gated calcium channel blockade with isradipine as a therapeutic strategy for Alzheimer’s disease. Neurobiol. Dis. 41, 62–70. doi: 10.1016/j.nbd.2010.08.020
Arendt, T., Stieler, J. T., and Holzer, M. (2016). Tau and tauopathies. Brain Res. Bull. 126, 238–292. doi: 10.1016/j.brainresbull.2016.08.018
Barnstedt, O., Owald, D., Felsenberg, J., Brain, R., Moszynski, J. P., Talbot, C. B., et al. (2016). Memory-Relevant mushroom body output synapses are cholinergic. Neuron 89, 1237–1247. doi: 10.1016/j.neuron.2016.02.015
Biundo, F., Del Prete, D., Zhang, H., Arancio, O., and D’Adamio, L. (2018). A role for tau in learning, memory and synaptic plasticity. Sci. Rep. 8:3184. doi: 10.1038/s41598-018-21596-3
Booth, C. A., Ridler, T., Murray, T. K., Ward, M. A., de Groot, E., Goodfellow, M., et al. (2016a). Electrical and network neuronal properties are preferentially disrupted in dorsal, but not ventral, medial entorhinal cortex in a mouse model of tauopathy. J. Neurosci. 36, 312–324. doi: 10.1523/JNEUROSCI.2845-14.2016
Booth, C. A., Witton, J., Nowacki, J., Tsaneva-Atanasova, K., Jones, M. W., Randall, A. D., et al. (2016b). Altered intrinsic pyramidal neuron properties and pathway-specific synaptic dysfunction underlie aberrant hippocampal network function in a mouse model of tauopathy. J. Neurosci. 36, 350–363. doi: 10.1523/JNEUROSCI.2151-15.2016
Boutajangout, A., Boom, A., Leroy, K., and Brion, J. P. (2004). Expression of tau mRNA and soluble tau isoforms in affected and non-affected brain areas in Alzheimer’s disease. FEBS Lett. 576, 183–189. doi: 10.1016/j.febslet.2004.09.011
Bowden, S. E., Fletcher, S., Loane, D. J., and Marrion, N. V. (2001). Somatic colocalization of rat SK1 and D class (Cav1.2) L-type calcium channels in rat CA1 hippocampal pyramidal neurons. 21:RC175. doi: 10.1523/JNEUROSCI.21-20-j0006.2001
Brzyska, M., and Elbaum, D. (2003). Dysregulation of calcium in Alzheimer’s disease. Acta Neurobiol. Exp. 63, 171–183.
Buee, L., Bussiere, T., Buee-Scherrer, V., Delacourte, A., and Hof, P. R. (2000). Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 33, 95–130. doi: 10.1016/s0165-0173(00)00019-9
Buhl, E., Higham, J. P., and Hodge, J. J. L. (2019). Alzheimer’s disease-associated tau alters Drosophila circadian activity, sleep and clock neuron electrophysiology. Neurobiol. Dis. 130:104507. doi: 10.1016/j.nbd.2019.104507
Canzoniero, L. M., and Snider, B. J. (2005). Calcium in Alzheimer’s disease pathogenesis: too much, too little or in the wrong place? J. Alzheimers Dis. 8, 147–154; discussion 209–115.
Cavaliere, S., Gillespie, J. M., and Hodge, J. J. (2012). KCNQ channels show conserved ethanol block and function in ethanol behaviour. PLoS One 7:e50279. doi: 10.1371/journal.pone.0050279
Cavaliere, S., Malik, B. R., and Hodge, J. J. (2013). KCNQ channels regulate age-related memory impairment. PLoS One 8:e62445. doi: 10.1371/journal.pone.0062445
Chee, F. C., Mudher, A., Cuttle, M. F., Newman, T. A., MacKay, D., Lovestone, S., et al. (2005). Over-expression of tau results in defective synaptic transmission in Drosophila neuromuscular junctions. Neurobiol. Dis. 20, 918–928. doi: 10.1016/j.nbd.2005.05.029
Coon, A. L., Wallace, D. R., Mactutus, C. F., and Booze, R. M. (1999). L-type calcium channels in the hippocampus and cerebellum of Alzheimer’s disease brain tissue. Neurobiol. Aging 20, 597–603. doi: 10.1016/s0197-4580(99)00068-8
Copenhaver, P. F., Anekonda, T. S., Musashe, D., Robinson, K. M., Ramaker, J. M., Swanson, T. L., et al. (2011). A translational continuum of model systems for evaluating treatment strategies in Alzheimer’s disease: isradipine as a candidate drug. Dis. Models Mech. 4, 634–648. doi: 10.1242/dmm.006841
Daschil, N., Kniewallner, K. M., Obermair, G. J., Hutter-Paier, B., Windisch, M., Marksteiner, J., et al. (2015). L-type calcium channel blockers and substance P induce angiogenesis of cortical vessels associated with beta-amyloid plaques in an Alzheimer mouse model. Neurobiol. Aging 36, 1333–1341. doi: 10.1016/j.neurobiolaging.2014.12.027
Davis, R. L. (2011). Traces of Drosophila memory. Neuron 70, 8–19. doi: 10.1016/j.neuron.2011.03.012
Dwight, Z., Palais, R., and Wittwer, C. T. (2011). uMELT: prediction of high-resolution melting curves and dynamic melting profiles of PCR products in a rich web application. Bioinformatics 27, 1019–1020. doi: 10.1093/bioinformatics/btr065
Espinoza, M., de Silva, R., Dickson, D. W., and Davies, P. (2008). Differential incorporation of tau isoforms in Alzheimer’s disease. J. Alzheimers Dis. 14, 1–16. doi: 10.3233/jad-2008-14101
Gillespie, J. M., and Hodge, J. J. (2013). CASK regulates CaMKII autophosphorylation in neuronal growth, calcium signaling, and learning. Front. Mol. Neurosci. 6:27. doi: 10.3389/fnmol.2013.00027
Goodison, W. V., Frisardi, V., and Kehoe, P. G. (2012). Calcium channel blockers and Alzheimer’s disease: potential relevance in treatment strategies of metabolic syndrome. J. Alzheimes Dis. 30(Suppl. 2), S269–S282. doi: 10.3233/JAD-2012-111664
Hasegawa, M., Watanabe, S., Kondo, H., Akiyama, H., Mann, D. M., Saito, Y., et al. (2014). 3R and 4R tau isoforms in paired helical filaments in Alzheimer’s disease. Acta Neuropathol. 127, 303–305. doi: 10.1007/s00401-013-1191-9
Higham, J. P., Malik, B. R., Buhl, E., Dawson, J. M., Ogier, A. S., Lunnon, K., et al. (2019). Alzheimer’s disease associated genes ankyrin and tau cause shortened lifespan and memory loss in Drosophila. Front. Cell. Neurosci. 13:260. doi: 10.3389/fncel.2019.00260
Iijima-Ando, K., and Iijima, K. (2010). Transgenic Drosophila models of Alzheimer’s disease and tauopathies. Brain Struct. Funct. 214, 245–262. doi: 10.1007/s00429-009-0234-4
Jiang, S. A., Campusano, J. M., Su, H., and O’Dowd, D. K. (2005). Drosophila mushroom body Kenyon cells generate spontaneous calcium transients mediated by PLTX-sensitive calcium channels. J. Neurophysiol. 94, 491–500. doi: 10.1152/jn.00096.2005
Kadas, D., Klein, A., Krick, N., Worrell, J. W., Ryglewski, S., and Duch, C. (2017). Dendritic and Axonal L-Type calcium channels cooperate to enhance motoneuron firing output during Drosophila larval locomotion. J. Neurosci. 37, 10971–10982. doi: 10.1523/JNEUROSCI.1064-17.2017
Kadas, D., Papanikolopoulou, K., Xirou, S., Consoulas, C., and Skoulakis, E. M. C. (2019). Human Tau isoform-specific presynaptic deficits in a Drosophila central nervous system circuit. Neurobiol. Dis. 124, 311–321. doi: 10.1016/j.nbd.2018.12.004
Kaufman, S. K., Del Tredici, K., Thomas, T. L., Braak, H., and Diamond, M. I. (2018). Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer’s disease and PART. Acta Neuropathol. 136, 57–67. doi: 10.1007/s00401-018-1855-6
Kerr, F., Augustin, H., Piper, M. D., Gandy, C., Allen, M. J., Lovestone, S., et al. (2011). Dietary restriction delays aging, but not neuronal dysfunction, in Drosophila models of Alzheimer’s disease. Neurobiol. Aging 32, 1977–1989. doi: 10.1016/j.neurobiolaging.2009.10.015
Kosmidis, S., Grammenoudi, S., Papanikolopoulou, K., and Skoulakis, E. M. (2010). Differential effects of Tau on the integrity and function of neurons essential for learning in Drosophila. J. Neurosci. 30, 464–477. doi: 10.1523/JNEUROSCI.1490-09.2010
Krashes, M. J., Keene, A. C., Leung, B., Armstrong, J. D., and Waddell, S. (2007). Sequential use of mushroom body neuron subsets during Drosophila odor memory processing. Neuron 53, 103–115. doi: 10.1016/j.neuron.2006.11.021
Lancaster, B., and Adams, P. R. (1986). Calcium-dependent current generating the afterhyperpolarization of hippocampal neurons. J. Neurophysiol. 55, 1268–1282. doi: 10.1152/jn.1986.55.6.1268
Malik, B. R., Gillespie, J. M., and Hodge, J. J. (2013). CASK and CaMKII function in the mushroom body alpha’/beta’ neurons during Drosophila memory formation. Front. Neural Circ. 7:52. doi: 10.3389/fncir.2013.00052
Malik, B. R., and Hodge, J. J. (2014). Drosophila adult olfactory shock learning. J. Vis. Exp. 90:e50107.
Marrion, N. V., and Tavalin, S. J. (1998). Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature 395, 900–905. doi: 10.1038/27674
Mattson, M. P., and Chan, S. L. (2001). Dysregulation of cellular calcium homeostasis in Alzheimer’s disease: bad genes and bad habits. J. Mol. Neurosci. 17, 205–224. doi: 10.1385/jmn:17:2:205
Mershin, A., Pavlopoulos, E., Fitch, O., Braden, B. C., Nanopoulos, D. V., and Skoulakis, E. M. (2004). Learning and memory deficits upon TAU accumulation in Drosophila mushroom body neurons. Learn Mem. 11, 277–287. doi: 10.1101/lm.70804
Missiaen, L., Robberecht, W., van den Bosch, L., Callewaert, G., Parys, J. B., Wuytack, F., et al. (2000). Abnormal intracellular Ca2+homeostasis and disease. Cell Calcium 28, 1–21. doi: 10.1054/ceca.2000.0131
Moosmang, S., Haider, N., Klugbauer, N., Adelsberger, H., Langwieser, N., Muller, J., et al. (2005). Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J. Neurosci. 25, 9883–9892. doi: 10.1523/jneurosci.1531-05.2005
Moyer, J. R. Jr., Power, J. M., Thompson, L. T., and Disterhoft, J. F. (2000). Increased excitability of aged rabbit CA1 neurons after trace eyeblink conditioning. J. Neurosci. 20, 5476–5482. doi: 10.1523/jneurosci.20-14-05476.2000
Moyer, J. R. Jr., Thompson, L. T., Black, J. P., and Disterhoft, J. F. (1992). Nimodipine increases excitability of rabbit CA1 pyramidal neurons in an age- and concentration-dependent manner. J. Neurophysiol. 68, 2100–2109. doi: 10.1152/jn.1992.68.6.2100
Nimmrich, V., and Eckert, A. (2013). Calcium channel blockers and dementia. Br. J. Pharmacol. 169, 1203–1210.
Nisbet, R. M., and Gotz, J. (2018). Amyloid-beta and Tau in Alzheimer’s Disease: novel pathomechanisms and non-pharmacological treatment strategies. J. Alzheimers Dis. 64, S517–S527. doi: 10.3233/JAD-179907
Nunez-Santana, F. L., Oh, M. M., Antion, M. D., Lee, A., Hell, J. W., and Disterhoft, J. F. (2014). Surface L-type Ca2+ channel expression levels are increased in aged hippocampus. Aging Cell 13, 111–120. doi: 10.1111/acel.12157
Owald, D., Felsenberg, J., Talbot, C. B., Das, G., Perisse, E., Huetteroth, W., et al. (2015). Activity of defined mushroom body output neurons underlies learned olfactory behavior in Drosophila. Neuron 86, 417–427. doi: 10.1016/j.neuron.2015.03.025
Papanikolopoulou, K., and Skoulakis, E. M. (2011). The power and richness of modelling tauopathies in Drosophila. Mol. Neurobiol. 44, 122–133. doi: 10.1007/s12035-011-8193-1
Pascual, A., and Preat, T. (2001). Localization of long-term memory within the Drosophila mushroom body. Science 294, 1115–1117. doi: 10.1126/science.1064200
Peng, I. F., and Wu, C. F. (2007). Drosophila cacophony channels: a major mediator of neuronal Ca2+ currents and a trigger for K+ channel homeostatic regulation. J. Neurosci. 27, 1072–1081. doi: 10.1523/jneurosci.4746-06.2007
Pitman, J. L., Huetteroth, W., Burke, C. J., Krashes, M. J., Lai, S. L., Lee, T., et al. (2011). A pair of inhibitory neurons are required to sustain labile memory in the Drosophila mushroom body. Curr. Biol. 21, 855–861. doi: 10.1016/j.cub.2011.03.069
Ryglewski, S., Lance, K., Levine, R. B., and Duch, C. (2012). Cav2 channels mediate low and high voltage-activated calcium currents in Drosophila motoneurons. J. Physiol. 590, 809–825. doi: 10.1113/jphysiol.2011.222836
Schlichting, M., Menegazzi, P., Lelito, K. R., Yao, Z., Buhl, E., Dalla Benetta, E., et al. (2016). A neural network underlying circadian entrainment and photoperiodic adjustment of sleep and activity in Drosophila. J. Neurosci. 36, 9084–9096. doi: 10.1523/JNEUROSCI.0992-16.2016
Sealey, M. A., Vourkou, E., Cowan, C. M., Bossing, T., Quraishe, S., Grammenoudi, S., et al. (2017). Distinct phenotypes of three-repeat and four-repeat human tau in a transgenic model of tauopathy. Neurobiol. Dis. 105, 74–83. doi: 10.1016/j.nbd.2017.05.003
Sejourne, J., Placais, P. Y., Aso, Y., Siwanowicz, I., Trannoy, S., Thoma, V., et al. (2011). Mushroom body efferent neurons responsible for aversive olfactory memory retrieval in Drosophila. Nat. Neurosci. 14, 903–910. doi: 10.1038/nn.2846
Shaw, R. E., Kottler, B., Ludlow, Z. N., Buhl, E., Kim, D., Morais da Silva, S., et al. (2018). In vivo expansion of functionally integrated GABAergic interneurons by targeted increase in neural progenitors. EMBO J. 37:e98163. doi: 10.15252/embj.201798163
Spillantini, M. G., and Goedert, M. (2013). Tau pathology and neurodegeneration. Lancet Neurol. 12, 609–622. doi: 10.1016/s1474-4422(13)70090-5
Spires-Jones, T. L., and Hyman, B. T. (2014). The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 82, 756–771. doi: 10.1016/j.neuron.2014.05.004
Straube, K. T., Deyo, R. A., Moyer, J. R. Jr., and Disterhoft, J. F. (1990). Dietary nimodipine improves associative learning in aging rabbits. Neurobiol. Aging 11, 659–661. doi: 10.1016/0197-4580(90)90033-v
Terada, S., Matsubara, D., Onodera, K., Matsuzaki, M., Uemura, T., and Usui, T. (2016). Neuronal processing of noxious thermal stimuli mediated by dendritic Ca2+ influx in Drosophila somatosensory neurons. eLife 5:e12959. doi: 10.7554/eLife.12959
Thibault, O., Gant, J. C., and Landfield, P. W. (2007). Expansion of the calcium hypothesis of brain aging and Alzheimer’s disease: minding the store. Aging Cell 6, 307–317. doi: 10.1111/j.1474-9726.2007.00295.x
Wang, Y., and Mattson, M. P. (2014). L-type Ca2+ currents at CA1 synapses, but not CA3 or dentate granule neuron synapses, are increased in 3xTgAD mice in an age-dependent manner. Neurobiol. Aging 35, 88–95. doi: 10.1016/j.neurobiolaging.2013.07.007
Wentzell, J., and Kretzschmar, D. (2010). Alzheimer’s disease and tauopathy studies in flies and worms. Neurobiol. Dis. 40, 21–28. doi: 10.1016/j.nbd.2010.03.007
White, J. A., McKinney, B. C., John, M. C., Powers, P. A., Kamp, T. J., and Murphy, G. G. (2008). Conditional forebrain deletion of the L-type calcium channel Cav1.2 disrupts remote spatial memories in mice. Learn. Mem. 15, 1–5. doi: 10.1101/lm.773208
Wittmann, C. W., Wszolek, M. F., Shulman, J. M., Salvaterra, P. M., Lewis, J., Hutton, M., et al. (2001). Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293, 711–714. doi: 10.1126/science.1062382
Worrell, J. W., and Levine, R. B. (2008). Characterization of voltage-dependent Ca2+ currents in identified Drosophila motoneurons in situ. J. Neurophysiol. 100, 868–878. doi: 10.1152/jn.90464.2008
Wu, J. W., Hussaini, S. A., Bastille, I. M., Rodriguez, G. A., Mrejeru, A., Rilett, K., et al. (2016). Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 19, 1085–1092. doi: 10.1038/nn.4328
Yu, D., Akalal, D. B., and Davis, R. L. (2006). Drosophila alpha/beta mushroom body neurons form a branch-specific, long-term cellular memory trace after spaced olfactory conditioning. Neuron 52, 845–855. doi: 10.1016/j.neuron.2006.10.030
Keywords: tau, tauopathy, Alzheimer’s disease, memory, L-type Ca2+ channels, Drosophila, GCaMP Ca2+ imaging
Citation: Higham JP, Hidalgo S, Buhl E and Hodge JJL (2019) Restoration of Olfactory Memory in Drosophila Overexpressing Human Alzheimer’s Disease Associated Tau by Manipulation of L-Type Ca2+ Channels. Front. Cell. Neurosci. 13:409. doi: 10.3389/fncel.2019.00409
Received: 29 March 2019; Accepted: 26 August 2019;
Published: 10 September 2019.
Edited by:
Miguel Medina, Biomedical Research Networking Center on Neurodegenerative Diseases (CIBERNED), SpainReviewed by:
Alfonso Martin-Pena, University of Florida, United StatesSe-Young Choi, Seoul National University, South Korea
Amrit Mudher, University of Southampton, United Kingdom
Copyright © 2019 Higham, Hidalgo, Buhl and Hodge. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James J. L. Hodge, amFtZXMuaG9kZ2VAYnJpc3RvbC5hYy51aw==
†These authors have contributed equally to this work